Polarization and finite electric fields¶
Polarization, and responses to finite electric fields for AlP.¶
This tutorial describes how to obtain the following physical properties, for an insulator:
- The polarization.
- The Born effective charge (by finite differences of polarization)
- The Born effective charge (by finite differences of forces)
- The dielectric constant
- The proper piezoelectric tensor (clamped and relaxed ions)
The case of the linear responses (for the Born effective charge, dielectric constant, piezoelectric tensor) is treated independently in other tutorials (Response-Function 1, Elastic properties), using Density-Functional Perturbation Theory. You will learn here how to obtain these quantities using finite difference techniques within ABINIT. To that end, we will describe how to compute the polarization, in the Berry phase formulation, and how to perform finite electric field calculations.
The basic theory for the Berry phase computation of the polarization was proposed by R. D. King-Smith and D. Vanderbilt in [Kingsmith1993]. The longer excellent paper by D. Vanderbilt and R. D. King-Smith ([Vanderbilt1993]) clarifies many aspects of this theory. Good overviews of this subject may be found in the review article [Resta1994] and book [Vanderbilt2018].
In order to gain the theoretical background needed to perform a calculation with a finite electric field, you should consider reading the following papers: [Souza2002], [Nunes2001] and M. Veithen PhD thesis. Finally, the extension to the PAW formalism specifically in ABINIT is discussed in [Gonze2009] and [Zwanziger2012].
This tutorial should take about 1 hour and 30 minutes.
Note
Supposing you made your own installation of ABINIT, the input files to run the examples are in the ~abinit/tests/ directory where ~abinit is the absolute path of the abinit top-level directory. If you have NOT made your own install, ask your system administrator where to find the package, especially the executable and test files.
In case you work on your own PC or workstation, to make things easier, we suggest you define some handy environment variables by executing the following lines in the terminal:
export ABI_HOME=Replace_with_absolute_path_to_abinit_top_level_dir # Change this line
export PATH=$ABI_HOME/src/98_main/:$PATH # Do not change this line: path to executable
export ABI_TESTS=$ABI_HOME/tests/ # Do not change this line: path to tests dir
export ABI_PSPDIR=$ABI_TESTS/Pspdir/ # Do not change this line: path to pseudos dir
Examples in this tutorial use these shell variables: copy and paste
the code snippets into the terminal (remember to set ABI_HOME first!) or, alternatively,
source the set_abienv.sh script located in the ~abinit directory:
source ~abinit/set_abienv.sh
The ‘export PATH’ line adds the directory containing the executables to your PATH so that you can invoke the code by simply typing abinit in the terminal instead of providing the absolute path.
To execute the tutorials, create a working directory (Work*) and
copy there the input files of the lesson.
Most of the tutorials do not rely on parallelism (except specific tutorials on parallelism). However you can run most of the tutorial examples in parallel with MPI, see the topic on parallelism.
1 Ground-state properties of AlP and general parameters¶
Before beginning, you might consider working in a different subdirectory, as for the other tutorials. For example, create Work_polarization in $ABI_TESTS/tutorespfn/Input
In this tutorial we will assume that the ground-state properties of AlP have been previously obtained, and that the corresponding convergence studies have been done. We will adopt the following set of generic parameters:
acell 3*7.2728565836E+00
ecut 5
ecutsm 0.5
dilatmx 1.05
nband 4 (=number of occupied bands)
ngkpt 6 6 6
nshiftk 4
shiftk 0.5 0.5 0.5
0.5 0.0 0.0
0.0 0.5 0.0
0.0 0.0 0.5
pseudopotentials Pseudodojo_nc_sr_04_pw_standard_psp8/P.psp8
Pseudodojo_nc_sr_04_pw_standard_psp8/Al.psp8
In principle, the acell to be used should be the one corresponding to the optimized structure at the ecut, and ngkpt combined with nshiftk and shiftk, chosen for the calculations.
For the purpose of this tutorial, in order to limit the duration of the runs, we are working at a low cutoff of 5 Ha, for which the optimized lattice constant is equal to \(7.27\times 2/\sqrt{2}=10.29~\mathrm{Bohr}\). Nonetheless this value is close to that obtained with a highly converged geometry optimization, of 10.25~Bohr. As always, if you adapt this tutorial to your own research, it is critical to perform full convergence studies. Before going further, you might refresh your memory concerning the other variables: ecutsm, dilatmx, ngkpt, nshiftk, and shiftk.
Note as well that the pseudopotentials used here are freely available from Pseudo Dojo. The ones chosen here for P and Al use the Perdew-Wang parameterization of the local density approximation (LDA); this is done to facilitate comparison of the results of this tutorial with those of Non-linear properties.
2 Berry phase calculation of polarization in zero field¶
In this section, you will learn how to perform a Berry phase calculation of the polarization. As a practical problem we will try to compute the Born effective charges from finite difference of the polarization (under finite atomic displacements), for AlP.
You can now copy the file tpolarization_1.abi to Work_polarization,
cd $ABI_TESTS/tutorespfn/Input
mkdir Workpolarization_
cd Workpolarization_
cp ../tpolarization_1.abi .
# Finite difference calculation of the Born effective charges of AlP # The calculation proceeds by computing the change in cell polarization # by a Berrys phase formula, due to moving one of the atoms in the cell # Berry phase calculation of the polarization # berryopt -1 triggers the implementation of M. Viethen berryopt -1 #Definition of the unit cell # these cell parameters were derived from a relaxation run done with the # current ecut and kpt values. The current ecut value used is very low but # is done to speed the calculations. # # Note that for a cubic space group as found here, the primitive unit # cell is often represented as (0,a/2,a/2); (a/2,0,a/2); (a/2,a/2,0). # Here abinit uses 1/sqrt(2) as the normalizer and adjusts acell (the cell # length scale) accordingly. All this is copied from the output of the relaxation # run. acell 7.2728565836E+00 7.2728565836E+00 7.2728565836E+00 Bohr rprim 0.0000000000E+00 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 0.0000000000E+00 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 0.0000000000E+00 #Definition of the atom types and pseudopotentials ntypat 2 # two types of atoms znucl 15 13 # the atom types are Phosphorous and Aluminum # the following norm-conserving pseudopotentials are stored in the abinit distribution, but are freely # available through www.pseudo-dojo.org # this set uses the Perdew-Wang parameterization of LDA for the xc model pp_dirpath "$ABI_PSPDIR" pseudos "Psdj_nc_sr_04_pw_std_psp8/P.psp8, Psdj_nc_sr_04_pw_std_psp8/Al.psp8" #Definition of the atoms natom 2 # two atoms in the cell typat 1 2 # type 1 is Phosphorous, type 2 is Aluminum (order defined by znucl above and pseudos list) nband 4 # nband is restricted here to the number of filled bands only, no empty bands. The theory of # the Berrys phase polarization formula assumes filled bands only. Our pseudopotential choice # includes 5 valence electrons on P, 3 on Al, for 8 total in the primitive unit cell, hence # 4 filled bands. #atomic positions. Here we do three separate computations, where the Al atom is moved (manually) #from its equilibrium position at 0,0,0 in calculation 1, to (+/-0.01,0,0) offsets in calculations #2 and 3. The P position is again taken from the value in the relaxation calculation done #previously. ndtset 3 jdtset 1 2 3 xcart1 2.5713431044E+00 2.5713431044E+00 2.5713431044E+00 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00 xcart2 2.5713431044E+00 2.5713431044E+00 2.5713431044E+00 1.0000000000E-02 0.0000000000E+00 0.0000000000E+00 xcart3 2.5713431044E+00 2.5713431044E+00 2.5713431044E+00 -1.0000000000E-02 0.0000000000E+00 0.0000000000E+00 #Numerical parameters of the calculation : planewave basis set and k point grid ecut 5 # this value is very low but is used here to achieve very low calculation times. # in a production environment this should be checked carefully for convergence and # a more reasonable value is probably around 20 ecutsm 0.5 dilatmx 1.05 ngkpt 6 6 6 nshiftk 4 # this Monkhorst-Pack shift pattern is used so that the symmetry of the shifted grid # is correct. A gamma-centered grid would also have the correct symmetry but would be # less efficient. shiftk 0.5 0.5 0.5 0.5 0.0 0.0 0.0 0.5 0.0 0.0 0.0 0.5 #Parameters for the SCF procedure toldfe 1.0d-15 nstep 8 # suppress printing of density, wavefunctions, etc for this tutorial prtwf 0 prtden 0 prteig 0 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% tpolarization_1.abo, tolnlines= 10, tolabs= 1.100e-08, tolrel= 3.000e-04 #%% [paral_info] #%% max_nprocs = 2 #%% [extra_info] #%% authors = J. Zwanziger, M. Veithen #%% keywords = NC, DFPT #%% description = #%% Finite difference calculation of the Born effective charges of AlP #%%<END TEST_INFO>
Note that two pseudopotentials are mentioned in this input file: one for the phosphorus atom, and one for the aluminum atom. The first listed, for P in this case, will define the first type of atom. The second listed, for Al, will define the second type of atom. This might be the first time that you encounter this situation (more than one type of atom) in the tutorials, in contrast with the four “basic” tutorials. Because of the use of more than one type of atom, the following input variables must be present:
You can start the calculation. It should take about 90 seconds on a typical desktop machine. In the meantime, examine the tpolarization_1.abi file. It includes three computations (see the section labelled as atomic positions) corresponding to, first, the reference optimized structure (\(\tau=0\)), and then to the structure with the Al atom displaced from 0.01 bohr to the right and to the left (referred to as \(\tau = +0.01\) and \(\tau =-0.01\)). These are typical for the amplitude of atomic displacement in this kind of finite difference computation. Notice also that the displacements are given using xcart, that is, explicitly in Cartesian directions in atomic units, rather than the primitive cell axes (which would use xred). This makes the correspondence with the polarization output in Cartesian directions much simpler to understand.
There are two implementations of the Berry phase within ABINIT. One is triggered by positive values of berryopt and was implemented by Na Sai. The other one is triggered by negative values of berryopt and was implemented by Marek Veithen. Both are suitable to compute the polarization, however, here we will focus on the implementation of Marek Veithen for two reasons. First, the results are directly provided in Cartesian coordinates at the end of the run (while the implementation of Na Sai reports them in reduced coordinates). Second, the implementation of Marek Veithen is the one to be used for the finite electric field calculation as described in the next section. Finally, note also that Veithen’s implementation works with kptopt = 1 or 2 while Na Sai implementation is restricted to kptopt = 2, which is less convenient.
The input file is typical for a self-consistent ground state calculation. In addition to the usual variables, for the Berry phase calculation we simply need to include berryopt (see also rfdir, the default being usually adequate):
berryopt -1
Make the run, then open the output file and look for the occurrence “Berry”. The output reports values of the Berry phase for individual k-point strings.
Computing the polarization (Berry phase) for reciprocal vector:
0.16667 0.00000 0.00000 (in reduced coordinates)
-0.01620 0.01620 0.01620 (in cartesian coordinates - atomic units)
Number of strings: 144
Number of k points in string: 6
Summary of the results
Electronic Berry phase 2.206976733E-03
Ionic phase -7.500000000E-01
Total phase -7.477930233E-01
Remapping in [-1,1] -7.477930233E-01
Polarization -1.632453164E-02 (a.u. of charge)/bohr^2
Polarization -9.340041842E-01 C/m^2
Other subtleties of Berry phases, explained in [Vanderbilt1993], also apply. First, note that neither the electronic Berry phase nor the ionic phase vanish in this highly symmetric case, contrary to intuition. Even though AlP does not have inversion symmetry, it does have tetrahedral symmetry, which would be enough to make an ordinary vector vanish. But a lattice-valued vector does not have to vanish: the lattice just has to transform into itself under the tetrahedral point group. The ionic phase corresponds actually to a lattice-valued vector (-¾ -¾ -¾). Concerning the electronic phase, it does not exactly vanish, unless the sampling of k points becomes continuous.
If you go further in the file you will find the final results in cartesian coordinates. You can collect them for the different values of \(\tau\).
\(\tau = 0\)
Polarization in cartesian coordinates (a.u.):
Total: -0.282749182E-01 -0.282749182E-01 -0.282749182E-01
Polarization in cartesian coordinates (C/m^2):
Total: -0.161774270E+01 -0.161774270E+01 -0.161774270E+01
Polarization in cartesian coordinates (a.u.):
Total: -0.281920467E-01 -0.282749119E-01 -0.282749119E-01
Polarization in cartesian coordinates (C/m^2):
Total: -0.161300123E+01 -0.161774234E+01 -0.161774234E+01
Polarization in cartesian coordinates (a.u.):
Total: -0.283577762E-01 -0.282749119E-01 -0.282749119E-01
Polarization in cartesian coordinates (C/m^2):
Total: -0.162248340E+01 -0.161774234E+01 -0.161774234E+01
For comparison, the same calculation using Density-Functional Perturbation Theory (DFPT) can be done by using the file $ABI_TESTS/tutorespfn/Input/tpolarization_2.abi.
# Linear response calculation for AlP # Perturbation: atomic displacements, electric fields, & strains # Finite difference calculation of the ddk # DFPT calculations require a series of computations to # to derive all necessary information ndtset 3 #DATASET1 : scf calculation: GS WF in the BZ #******************************************** prtden1 1 prtwf1 1 kptopt1 1 tolvrs1 1.0D-18 nstep1 8 #DATASET2 : non scf calculation: GS WF in the whole BZ #***************************************************** getden2 1 kptopt2 2 iscf2 -2 getwfk2 1 tolwfr2 1.0d-22 berryopt2 -2 # berryopt -2 provides the DDK perturbation through a finite # difference formula, roughly |du/dk> = (|u_k+dk> - |u_k-dk>)/(2*dk) # It would also be possible to use rfddk 1 here, and compute the DDK # wavefunctions from within the DFPT formalism prtwf2 1 #DATASET3 : linear response to atomic displacements #************************************************** getwfk3 2 rfphon3 1 rfstrs3 3 rfelfd3 3 getddk3 2 tolvrs3 1.0d-12 kptopt3 2 nstep3 8 #Definition of the unit cell # these cell parameters were derived from a relaxation run done with the # current ecut and kpt values. The current ecut value used is very low but # is done to speed the calculations. # acell 7.2728565836E+00 7.2728565836E+00 7.2728565836E+00 Bohr rprim 0.0000000000E+00 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 0.0000000000E+00 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 0.0000000000E+00 #Definition of the atom types and pseudopotentials ntypat 2 # two types of atoms znucl 15 13 # the atom types are Phosphorous and Aluminum pp_dirpath "$ABI_PSPDIR" pseudos "Psdj_nc_sr_04_pw_std_psp8/P.psp8, Psdj_nc_sr_04_pw_std_psp8/Al.psp8" #Definition of the atoms natom 2 # two atoms in the cell typat 1 2 # type 1 is Phosphorous, type 2 is Aluminum (order defined by znucl above and pseudos list) nband 4 # nband is restricted here to the number of filled bands only, no empty bands. #atomic positions. xred # atomic positions are given in units of the primitive cell vectors, as is common # in crystallography 1/4 1/4 1/4 # P position 0 0 0 # Al position #Numerical parameters of the calculation : planewave basis set and k point grid ecut 5 # this value is very low but is used here to achieve very low calculation times. # in a production environment this should be checked carefully for convergence and # a more reasonable value is probably around 20 ecutsm 0.5 ngkpt 6 6 6 nshiftk 4 # this Monkhorst-Pack shift pattern is used so that the symmetry of the shifted grid # is correct. A gamma-centered grid would also have the correct symmetry but would be # less efficient. shiftk 0.5 0.5 0.5 0.5 0.0 0.0 0.0 0.5 0.0 0.0 0.0 0.5 # by default, don't save files, only save files within each # data set as needed prtwf 0 prtden 0 prteig 0 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = tpolarization_2.abi, tpolarization_3.abi #%% [files] #%% files_to_test = #%% tpolarization_2.abo, tolnlines= 2, tolabs= 5.000e-07, tolrel= 3.000e-04, fld_options=-medium #%% [paral_info] #%% max_nprocs = 2 #%% [extra_info] #%% authors = J. Zwanziger, M. Veithen #%% keywords = NC, DFPT #%% description = #%% Linear response calculation for AlP #%% Perturbation: atomic displacements, strains, electric fields #%% Finite difference calculation of the ddk #%%<END TEST_INFO>
Actually, the file tpolarization_2.abi not only leads to the computation of the Born effective charges, but also the computation of the piezoelectric constants (see later). You can review how to use DFPT in the tutorial Response-function 1 and tutorial Response-function 2 tutorials.
Note
An interesting feature of tpolarization_2.abi is the use of berryopt2 -2 in
the second data set. This input variable causes the computation of the DDK
wavefunctions using a finite difference formula, rather than the DFPT approach
triggered by rfddk. Although not strictly required in the present DFPT calculation,
the finite difference approach is necessary in the various
Berry’s phase computations of polarization, in order to maintain phase coherency
between wavefunctions at neighboring k points. Therefore in the present tutorial we use
the finite difference approach, in order to compare the results of the Berry’s phase
computation to those of DFPT more accurately.
Warning
The use of kpoint overlaps in Berry’s phase calculations is necessary, but causes the results to converge much more slowly with kpoint mesh density than other types of calculations. It is critical in production work using Berry’s phase methods to check carefully the convergence with respect to kpoint mesh density.
Go ahead and run the input file, and have a look at the output file, to identify the place where the Born effective charge is written (search for the phrase “Effective charges”). The value we get from DFPT is 2.254, in surprisingly good agreement with the above-mentioned value of 2.258. This level of agreement is fortuitous for unconverged calculations. Both methods (finite-difference and DFPT) will tend to the same value for better converged calculations.
The DDB file generated by $ABI_TESTS/tutorespfn/Input/tpolarization_2.abi can be used as input to anaddb for further processing, using the input file tpolarization_3.abi and the tpolarization_3.files file.
tffield_3.abi tffield_3.abo tffield_2o_DS3_DDB tffield_3_band2eps tffield_dummy1 tffield_dummy2 tffield_dummy3
# anaddb input file to compute DFPT properties of AlP, # based on previous abinit computations in tpolarization_2.abi # General information #******************** rfmeth 1 chneut 2 # Flags #******* elaflag 3 piezoflag 3 instrflag 1 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = anaddb #%% test_chain = tpolarization_2.abi, tpolarization_3.abi #%% input_ddb = tpolarization_2o_DS3_DDB #%% [files] #%% files_to_test = #%% tpolarization_3.abo, tolnlines= 2, tolabs= 5.000e-07, tolrel= 3.000e-04, fld_options=-medium #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = J. Zwanziger, M. Veithen, P. Ghosez #%% keywords = #%% description = Anaddb input file. #%%<END TEST_INFO>
Note
While the abinit program itself takes its input file as an
argument, the anaddb post-processing program depends in general on multiple
input files, and therefore it is more convenient to pipe in a file whose
contents are just the names of the files that anaddb should ultimately use. In
the present case, the piped-in file tpolarization_3.files is written such that the
DDB file is named tpolarization_2o_DS3_DDB (this is defined in the third line of
tpolarization_3.files). In the event of a mismatch, you can either edit
tpolarization_3.files to match the DDB you have, or change the name of the DDB
file.
The DFPT calculation yields the following piezoelectric constants, as found in tpolarization_3.abo:
Proper piezoelectric constants (clamped ion) (unit:c/m^2)
0.00000000 -0.00000000 0.00000000
0.00000000 0.00000000 0.00000000
-0.00000000 -0.00000000 -0.00000000
-0.64263948 0.00000000 0.00000000
0.00000000 -0.64263948 0.00000000
0.00000000 0.00000000 -0.64263948
....
Proper piezoelectric constants (relaxed ion) (unit:c/m^2)
0.00000000 0.00000000 -0.00000000
0.00000000 -0.00000000 -0.00000000
0.00000000 -0.00000000 -0.00000000
0.13114427 0.00000000 -0.00000000
0.00000000 0.13114427 -0.00000000
-0.00000000 -0.00000000 0.13114427
.Version 10.5.8.2 of ANADDB, released Oct 2025.
.(MPI version, prepared for a x86_64_linux_gnu13.2 computer)
.Copyright (C) 1998-2026 ABINIT group .
ANADDB comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Sat 20 Dec 2025.
- ( at 17h04 )
================================================================================
-outvars_anaddb: echo values of input variables ----------------------
Flags :
elaflag 3
instrflag 1
piezoflag 3
Miscellaneous information :
asr 1
chneut 2
================================================================================
read the DDB information and perform some checks
==== Info on the Cryst% object ====
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 5.1426862 5.1426862 G(1)= -0.0972255 0.0972255 0.0972255
R(2)= 5.1426862 0.0000000 5.1426862 G(2)= 0.0972255 -0.0972255 0.0972255
R(3)= 5.1426862 5.1426862 0.0000000 G(3)= 0.0972255 0.0972255 -0.0972255
Unit cell volume ucvol= 2.7201952E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Time-reversal symmetry is present
Reduced atomic positions [iatom, xred, symbol]:
1) 0.2500000 0.2500000 0.2500000 P
2) 0.0000000 0.0000000 0.0000000 Al
DDB file with 1 blocks has been read.
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 0.000065 0.000000
1 2 -0.000000 0.000000
1 3 -0.000000 0.000000
2 1 -0.000000 0.000000
2 2 0.000065 0.000000
2 3 0.000000 0.000000
3 1 0.000000 0.000000
3 2 0.000000 0.000000
3 3 0.000065 0.000000
Effective charge tensors after
imposition of the charge neutrality (if requested by user),
and eventual restriction to some part :
atom displacement
1 1 -2.254053E+00 -2.101373E-17 -2.096658E-17
1 2 -2.101373E-17 -2.254053E+00 2.106087E-17
1 3 2.101373E-17 2.101373E-17 -2.254053E+00
2 1 2.254053E+00 2.101373E-17 2.096658E-17
2 2 2.101373E-17 2.254053E+00 -2.106087E-17
2 3 -2.101373E-17 -2.101373E-17 2.254053E+00
Now, the imaginary part of the dynamical matrix is zeroed
================================================================================
Calculation of the internal-strain tensor
Force-response internal strain tensor(Unit:Hartree/bohr)
Atom dir strainxx strainyy strainzz strainyz strainxz strainxy
1 x -0.0000000 -0.0000000 -0.0000000 -0.1729192 -0.0000000 0.0000000
1 y -0.0000000 -0.0000000 -0.0000000 -0.0000000 -0.1729192 0.0000000
1 z 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 -0.1729192
2 x 0.0000000 0.0000000 0.0000000 0.1729192 0.0000000 -0.0000000
2 y 0.0000000 0.0000000 0.0000000 0.0000000 0.1729192 -0.0000000
2 z -0.0000000 -0.0000000 -0.0000000 -0.0000000 -0.0000000 0.1729192
Displacement-response internal strain tensor (Unit:Bohr)
Atom dir strainxx strainyy strainzz strainyz strainxz strainxy
1 x -0.0000000 -0.0000000 -0.0000000 -0.8160179 -0.0000000 0.0000000
1 y -0.0000000 -0.0000000 -0.0000000 -0.0000000 -0.8160179 0.0000000
1 z 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 -0.8160179
2 x 0.0000000 0.0000000 0.0000000 0.8160179 0.0000000 -0.0000000
2 y 0.0000000 0.0000000 0.0000000 0.0000000 0.8160179 -0.0000000
2 z -0.0000000 -0.0000000 -0.0000000 -0.0000000 -0.0000000 0.8160179
================================================================================
Calculation of the elastic and compliances tensor (Voigt notation)
Elastic Tensor (clamped ion) (unit:10^2GP):
1.4315873 0.7331581 0.7331581 0.0000000 0.0000000 -0.0000000
0.7331581 1.4315873 0.7331581 0.0000000 0.0000000 -0.0000000
0.7331581 0.7331581 1.4315873 0.0000000 0.0000000 -0.0000000
-0.0000000 -0.0000000 -0.0000000 0.9802774 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.9802774 -0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 -0.0000000 0.9802774
Elastic Tensor (relaxed ion) (unit:10^2GP):
(at fixed electric field boundary condition)
1.4315873 0.7331581 0.7331581 0.0000000 0.0000000 -0.0000000
0.7331581 1.4315873 0.7331581 0.0000000 0.0000000 -0.0000000
0.7331581 0.7331581 1.4315873 0.0000000 0.0000000 -0.0000000
-0.0000000 -0.0000000 -0.0000000 0.6750452 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.6750452 -0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 -0.0000000 0.6750452
Compliance Tensor (clamped ion) (unit: 10^-2GP^-1):
1.0695486 -0.3622358 -0.3622358 -0.0000000 -0.0000000 0.0000000
-0.3622358 1.0695486 -0.3622357 -0.0000000 -0.0000000 0.0000000
-0.3622358 -0.3622357 1.0695486 -0.0000000 -0.0000000 0.0000000
-0.0000000 0.0000000 0.0000000 1.0201194 -0.0000000 -0.0000000
-0.0000000 0.0000000 0.0000000 -0.0000000 1.0201194 0.0000000
-0.0000000 -0.0000000 0.0000000 -0.0000000 0.0000000 1.0201194
Compliance Tensor (relaxed ion) (unit: 10^-2GP^-1):
(at fixed electric field boundary condition)
1.0695486 -0.3622358 -0.3622358 -0.0000000 -0.0000000 0.0000000
-0.3622358 1.0695486 -0.3622357 -0.0000000 -0.0000000 0.0000000
-0.3622358 -0.3622357 1.0695486 -0.0000000 -0.0000000 0.0000000
-0.0000000 0.0000000 0.0000000 1.4813823 -0.0000000 -0.0000000
-0.0000000 0.0000000 0.0000000 -0.0000000 1.4813823 0.0000000
-0.0000000 -0.0000000 0.0000000 -0.0000000 0.0000000 1.4813823
================================================================================
Calculation of the tensor related to piezoelectric effetc
(Elastic indices in Voigt notation)
Proper piezoelectric constants (clamped ion) (unit:c/m^2)
0.00000000 0.00000000 -0.00000000
0.00000000 0.00000000 0.00000000
0.00000000 0.00000000 0.00000000
-0.64263952 0.00000000 0.00000000
0.00000000 -0.64263952 0.00000000
0.00000000 0.00000000 -0.64263952
ddb_piezo : WARNING -
Acoustic sum rule violation met : the eigenvalues of accoustic mode
are too large at Gamma point
Increase cutoff energy or k-points sampling.
The three eigenvalues are: -8.771287E-06 -3.152799E-17 -8.771287E-06
Proper piezoelectric constants (relaxed ion) (unit:c/m^2)
0.00000000 0.00000000 -0.00000000
0.00000000 0.00000000 -0.00000000
0.00000000 0.00000000 -0.00000000
0.13114445 0.00000000 -0.00000000
0.00000000 0.13114445 -0.00000000
-0.00000000 -0.00000000 0.13114445
-
- Proc. 0 individual time (sec): cpu= 0.0 wall= 0.0
================================================================================
+Total cpu time 0.034 and wall time 0.034 sec
anaddb : the run completed successfully.
The piezoelectric constants here are the change in polarization as a function of strain [Wu2005]. The rows are the strain directions using Voigt notation (directions 1-6) while the columns are the polarization directions. In the \(\bar{4}3m\) crystal class of AlP, the only non-zero piezoelectric elements are those associated with shear strain (Voigt notation strains \(e_4\), \(e_5\), and \(e_6\)) [Nye1985].
The relaxed ion values, where the ionic relaxation largely suppresses the electronic piezoelectricity, will be more difficult to converge than the clamped ion result.
Because the Berry phase approach computes polarization, it can also be used to compute the piezoelectric constants from finite difference of polarization with respect to strains. This can be done considering clamped ions or relaxed ions configurations. For this purpose, have a look at the files t_polarization4.abi (clamped ions) and t_polarization5.abi (relaxed ions).
# Finite difference calculation of the clamped-ion # piezoelectric constants of AlP # # The calculation proceeds by computing the change in cell # polarization by a Berrys phase formula, due to changes # in the strain of the unit cell. "Clamped-ion" refers to the # fact that the ions move with the strains but are not # allowed to relax to their new equilibrium positions # Berry phase calculation of the polarization # berryopt -1 triggers the implementation of M. Viethen berryopt -1 # three cell geometries will be computed ndtset 3 #Definition of the unit cell acell 3*7.2728565836E+00 # rprim = strain tensor x rprim0, where # rprim0 is the unstrained rprim # strain: e4 = 0.00 rprim1 0.0000000000E+00 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 0.0000000000E+00 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 0.0000000000E+00 #strain: e4 = 0.01 rprim2 0.0000000000E+00 7.1064231509E-01 7.1064231509E-01 7.0710678119E-01 3.535533906E-03 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 3.535533906E-03 # strain: e4 = -0.01 rprim3 0.0000000000E+00 7.0357124728E-01 7.0357124728E-01 7.0710678119E-01 -3.535533906E-03 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 -3.535533906E-03 tolsym 1.0e-8 # Since we make rather small lattice vector changes, with the default value of tolsym # ABINIT will recognize that we are close to a more symmetric # configuration, and will resymmetrize the primitive vectors, something we want to avoid ... #Definition of the atom types and pseudopotentials ntypat 2 # two types of atoms znucl 15 13 # the atom types are Phosphorous and Aluminum # the following norm-conserving pseudopotentials are stored in the abinit distribution, but are freely # available through www.pseudo-dojo.org # this set uses the Perdew-Wang parameterization of LDA for the xc model pp_dirpath "$ABI_PSPDIR" pseudos "Psdj_nc_sr_04_pw_std_psp8/P.psp8, Psdj_nc_sr_04_pw_std_psp8/Al.psp8" #Definition of the atoms natom 2 # two atoms in the cell typat 1 2 # type 1 is Phosphorous, type 2 is Aluminum (order defined by znucl above and pseudos list) nband 4 # nband is restricted here to the number of filled bands only, no empty bands. The theory of # the Berrys phase polarization formula assumes filled bands only. Our pseudopotential choice # includes 5 valence electrons on P, 3 on Al, for 8 total in the primitive unit cell, hence # 4 filled bands. #atomic positions, given in units of the cell vectors. Thus as the cell vectors #change due to strain the atoms will move as well. xred 1/4 1/4 1/4 0 0 0 #Numerical parameters of the calculation : planewave basis set and k point grid ecut 5 # this value is very low but is used here to achieve very low calculation times. # in a production environment this should be checked carefully for convergence and # a more reasonable value is probably around 20 ecutsm 0.5 dilatmx 1.05 ngkpt 6 6 6 nshiftk 4 # this Monkhorst-Pack shift pattern is used so that the symmetry of the shifted grid # is correct. A gamma-centered grid would also have the correct symmetry but would be # less efficient. shiftk 0.5 0.5 0.5 0.5 0.0 0.0 0.0 0.5 0.0 0.0 0.0 0.5 #Parameters for the SCF procedure toldfe 1.0d-15 nstep 8 # suppress printing of density, wavefunctions, etc for this tutorial prtwf 0 prtden 0 prteig 0 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% tpolarization_4.abo, tolnlines= 10, tolabs= 9.999e-9, tolrel= 1.224e-04 #%% [paral_info] #%% max_nprocs = 2 #%% [extra_info] #%% authors = J. Zwanziger, M. Veithen #%% keywords = NC, DFPT #%% description = #%% Finite difference calculation of the clamped-ion #%% piezoelectric constants of AlP #%%<END TEST_INFO>
# Finite difference calculation of the relaxed ion # piezoelectric constants of AlP # # The calculation proceeds by computing the change in cell # polarization by a Berrys phase formula, due to changes # in the strain of the unit cell. In this case, the ion positions # will be allowed to relax to to their new equilibrium positions # within the strain cell. # Berry phase calculation of the polarization # berryopt -1 triggers the implementation of M. Viethen berryopt -1 # three cell geometries will be computed ndtset 3 #Definition of the unit cell acell 3*7.2728565836E+00 # rprim = strain tensor x rprim0, where # rprim0 is the unstrained rprim # strain: e4 = 0.00 rprim1 0.0000000000E+00 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 0.0000000000E+00 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 0.0000000000E+00 #strain: e4 = 0.01 rprim2 0.0000000000E+00 7.1064231509E-01 7.1064231509E-01 7.0710678119E-01 3.535533906E-03 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 3.535533906E-03 # strain: e4 = -0.01 rprim3 0.0000000000E+00 7.0357124728E-01 7.0357124728E-01 7.0710678119E-01 -3.535533906E-03 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 -3.535533906E-03 tolsym 1.0e-8 # Since we make rather small lattice vector changes, with the default value of tolsym # ABINIT will recognize that we are close to a more symmetric # configuration, and will resymmetrize the primitive vectors, something we want to avoid ... #Definition of the atom types and pseudopotentials ntypat 2 # two types of atoms znucl 15 13 # the atom types are Phosphorous and Aluminum # the following norm-conserving pseudopotentials are stored in the abinit distribution, but are freely # available through www.pseudo-dojo.org # this set uses the Perdew-Wang parameterization of LDA for the xc model pp_dirpath "$ABI_PSPDIR" pseudos "Psdj_nc_sr_04_pw_std_psp8/P.psp8, Psdj_nc_sr_04_pw_std_psp8/Al.psp8" #Definition of the atoms natom 2 # two atoms in the cell typat 1 2 # type 1 is Phosphorous, type 2 is Aluminum (order defined by znucl above and pseudos list) nband 4 # nband is restricted here to the number of filled bands only, no empty bands. The theory of # the Berrys phase polarization formula assumes filled bands only. Our pseudopotential choice # includes 5 valence electrons on P, 3 on Al, for 8 total in the primitive unit cell, hence # 4 filled bands. #atomic positions, given in units of the cell vectors. Thus as the cell vectors #change due to strain the atoms will move as well. xred 1/4 1/4 1/4 0 0 0 # Relaxation of atomic positions optcell 0 # the cell geometry is not allowed to changed geoopt "bfgs" # the ion positions will be relaxed tolmxf 1.0d-5 ntime 13 #Numerical parameters of the calculation : planewave basis set and k point grid ecut 5 # this value is very low but is used here to achieve very low calculation times. # in a production environment this should be checked carefully for convergence and # a more reasonable value is probably around 20 ecutsm 0.5 dilatmx 1.05 ngkpt 6 6 6 nshiftk 4 # this Monkhorst-Pack shift pattern is used so that the symmetry of the shifted grid # is correct. A gamma-centered grid would also have the correct symmetry but would be # less efficient. shiftk 0.5 0.5 0.5 0.5 0.0 0.0 0.0 0.5 0.0 0.0 0.0 0.5 #Parameters for the SCF procedure tolvrs 1.0D-19 nstep 8 # suppress printing of density, wavefunctions, etc for this tutorial prtwf 0 prtden 0 prteig 0 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% tpolarization_5.abo, tolnlines= 22, tolabs= 1.100e-08, tolrel= 4.500e-04 #%% [paral_info] #%% max_nprocs = 2 #%% [extra_info] #%% authors = J. Zwanziger, M. Veithen #%% keywords = NC, DFPT #%% description = #%% Finite difference calculation of the clamped-ion #%% piezoelectric constants of AlP #%%<END TEST_INFO>
In these input files the finite strain is applied by multiplying the \(e_4\) (Voigt notation) strain tensor by the (dimensionless) unit cell vectors: $$ \left[\begin{matrix} 1 & 0 & 0 \\ 0 & 1 & e_4/2 \\ 0 & e_4/2 & 1 \\ \end{matrix}\right] \left[\begin{matrix} 0 & 1/\sqrt{2} & 1/\sqrt{2} \\ 1/\sqrt{2} & 0 & 1/\sqrt{2} \\ 1/\sqrt{2} & 1/\sqrt{2} & 0 \\ \end{matrix}\right] = \left[\begin{matrix} 0 & 1/\sqrt{2} & 1/\sqrt{2} \\ (1+e_4/2)/\sqrt{2} & e_4/2\sqrt{2} & 1/\sqrt{2} \\ (1+e_4/2)/\sqrt{2} & 1/\sqrt{2} & e_4/2\sqrt{2} \\ \end{matrix}\right] $$ Don’t forget that in the input file, vectors are read in as rows of numbers, not columns!
Notice how in the relaxed ion case, the input file includes geoopt “bfgs” and optcell = 0, in order to relax the ion positions at fixed cell geometry. These calculations should give the following final results (obtained by taking finite difference expressions of the strains for different electric fields): \(-0.6427~C/m^2\) for the clamped ion case, and \(0.1310~C/m^2\) for the relaxed ion case.
For example, the clamped ion piezoelectric constant was obtained from tpolarization_4.abo:
== DATASET 2 ==========================================================
....
Polarization in cartesian coordinates (C/m^2):
Total: -0.162420887E+01 -0.162587046E+01 -0.162587046E+01
....
== DATASET 3 ==========================================================
...
Polarization in cartesian coordinates (C/m^2):
Total: -0.161135421E+01 -0.160969264E+01 -0.160969264E+01
3 Finite electric field calculations¶
In this section, you will learn how to perform a calculation with a finite electric field.
You can now copy the file $ABI_TESTS/tutorespfn/Input/tpolarization_6.abi to Work_polarization.
# Finite electric field calculation of AlP at clamped atomic positions # # Here the polarization of the cell is computed as a function of increasing # external homogeneous electric field. # berryopt 4 is used to trigger the finite field calculation, while # the efield variable sets the strength (in atomic units) and direction # of the field # we can run many (11) fields, or just 3, to make a quick run. # ndtset 11 ndtset 3 jdtset 11 21 # 22 23 24 25 # The additional 8 values of the field have been suppressed to save CPU time 31 # 32 33 34 35 # the initial run is at zero field and uses berryopt -1 berryopt11 -1 # runs with finite field use berryopt 4, efield, and # must read in the wavefunctions of the previous run with smaller field # if variables tagged by numbers like 22, 23, 24, 25 are defined but # not called by jdtset (above), they will be quietly ignored. This # feature gives a lot of flexibility in an input file. berryopt21 4 efield21 0.0001 0.0001 0.0001 getwfk21 11 berryopt22 4 efield22 0.0002 0.0002 0.0002 getwfk22 21 berryopt23 4 efield23 0.0003 0.0003 0.0003 getwfk23 22 berryopt24 4 efield24 0.0004 0.0004 0.0004 getwfk24 23 berryopt25 4 efield25 0.0005 0.0005 0.0005 getwfk25 24 berryopt31 4 efield31 -0.0001 -0.0001 -0.0001 getwfk31 11 berryopt32 4 efield32 -0.0002 -0.0002 -0.0002 getwfk32 31 berryopt33 4 efield33 -0.0003 -0.0003 -0.0003 getwfk33 32 berryopt34 4 efield34 -0.0004 -0.0004 -0.0004 getwfk34 33 berryopt35 4 efield35 -0.0005 -0.0005 -0.0005 getwfk35 34 #Definition of the unit cell acell 3*7.2728565836E+00 rprim 0.0000000000E+00 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 0.0000000000E+00 7.0710678119E-01 7.0710678119E-01 7.0710678119E-01 0.0000000000E+00 #Definition of the atom types and pseudopotentials ntypat 2 # two types of atoms znucl 15 13 # the atom types are Phosphorous and Aluminum # the following norm-conserving pseudopotentials are stored in the abinit distribution, but are freely # available through www.pseudo-dojo.org # this set uses the Perdew-Wang parameterization of LDA for the xc model pp_dirpath "$ABI_PSPDIR" pseudos "Psdj_nc_sr_04_pw_std_psp8/P.psp8, Psdj_nc_sr_04_pw_std_psp8/Al.psp8" #Definition of the atoms natom 2 # two atoms in the cell typat 1 2 # type 1 is Phosphorous, type 2 is Aluminum (order defined by znucl above and pseudos list) nband 4 # nband is restricted here to the number of filled bands only, no empty bands. The theory of # the Berrys phase polarization formula assumes filled bands only. Our pseudopotential choice # includes 5 valence electrons on P, 3 on Al, for 8 total in the primitive unit cell, hence # 4 filled bands. #atomic positions, given in units of the cell vectors. Thus as the cell vectors #change due to strain the atoms will move as well. xred 1/4 1/4 1/4 0 0 0 #Numerical parameters of the calculation : planewave basis set and k point grid ecut 5 # this value is very low but is used here to achieve very low calculation times. # in a production environment this should be checked carefully for convergence and # a more reasonable value is probably around 20 ecutsm 0.5 dilatmx 1.05 ngkpt 6 6 6 nshiftk 4 # this Monkhorst-Pack shift pattern is used so that the symmetry of the shifted grid # is correct. A gamma-centered grid would also have the correct symmetry but would be # less efficient. shiftk 0.5 0.5 0.5 0.5 0.0 0.0 0.0 0.5 0.0 0.0 0.0 0.5 #Parameters for the SCF procedure nstep 7 toldfe 1.0d-15 # suppress printing of density and eigenvalues for this tutorial, # but we do need the wavefunctions prtwf 1 prtden 0 prteig 0 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% tpolarization_6.abo, tolnlines = 0, tolabs = 0.0e+00, tolrel = 0.0e+00, fld_options = -ridiculous #%% [paral_info] #%% max_nprocs = 2 #%% [extra_info] #%% authors = J. Zwanziger, M. Veithen #%% keywords = NC, DFPT #%% description = Finite electric field calculation of AlP at clamped atomic positions #%%<END TEST_INFO>
You can start the run immediately. It performs a finite field calculation at clamped atomic positions. You can look at this input file to identify the features specific to the finite field calculation.
As general parameters, one has to specify nband, nbdbuf and kptopt:
nband 4
nbdbuf 0
kptopt 1
As a first step (dataset 11), the code must perform a Berry phase calculation in zero electric field. For that purpose, it is necessary to set the values of berryopt:
berryopt11 -1
Warning
You cannot use berryopt to +1 to initiate a finite field calculation. You must begin with berryopt -1.
After that, there are different steps corresponding to various values of the electric field, as set by efield. For those steps, it is important to take care of the following parameters, as shown here for example for dataset 21:
berryopt21 4
efield21 0.0001 0.0001 0.0001
getwfk21 11
The electric field is applied here along the [111] direction. It must be incremented step by step (it is not possible to go to high field directly). At each step, the wavefunctions of the previous step must be used. When the field gets large, it is important to check that it does not significantly exceed the critical field for Zener breakthrough (the drop of potential over the Born-von Karmann supercell must be smaller than the gap). In practice, the number of k-point must be enlarged to reach convergence. However, at the same time, the critical field becomes smaller. In practice, reasonable fields can still be reached for k-point grids providing a reasonable degree of convergence. A compromise must however be found.
As these calculations are quite long, the input file has been limited to a small number of small fields. Three cases have been selected: \(E = 0\), \(E = +0.0001\) and \(E = -0.0001\). If you have time later, you can perform more exhaustive calculations over a larger set of fields.
Various quantities can be extracted from the finite field calculation at clamped ions using finite difference techniques: the Born effective charge \(Z^*\) can be extracted from the difference in forces at different electric fields, the optical dielectric constant can be deduced from the polarizations at different fields, and the clamped ion piezoelectric tensor can be deduced from the stress tensor at different fields. As an illustration we will focus here on the computation of \(Z^*\).
Examine the output file. For each field, the file contains various quantities that can be collected. For the present purpose, we can look at the evolution of the forces with the field and extract the following results from the output file:
\(E=0\)
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
2 -0.00000000000000 -0.00000000000000 -0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00022532204220 -0.00022532204220 -0.00022532204220
2 0.00022532204220 0.00022532204220 0.00022532204220
cartesian forces (hartree/bohr) at end:
1 0.00022548273033 0.00022548273033 0.00022548273033
2 -0.00022548273033 -0.00022548273033 -0.00022548273033
The value for positive and negative fields above are nearly the same, showing that the quadratic term is almost negligible. This clearly appears in the figure below where the field dependence of the force for a larger range of electric fields is plotted.
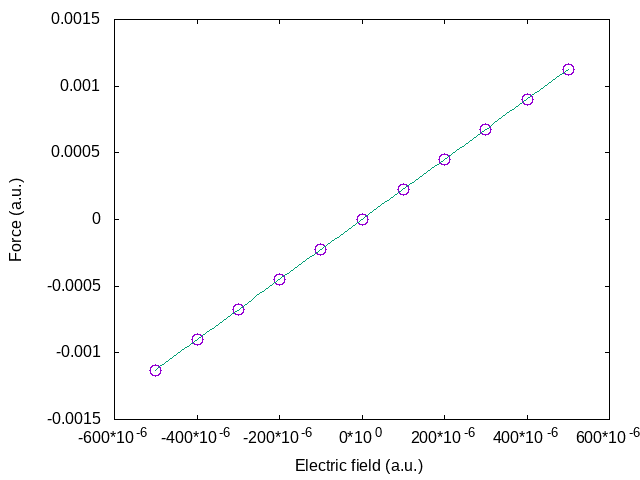
We can therefore extract with good accuracy the Born effective charge as:
This value is similar to the value reported from DFPT. If you do calculations for more electric fields, fitting them with the general expression of the force above (including the \(E^2\) term), you can find the \(d\chi/d\tau\) term. From the given input file tpolarization_6.abi, using all the fields, you should find \(d\chi/d\tau\) for Al of = -0.0295.
Going back to the output file, you can also look at the evolution of the polarization with the field.
\(E = 0\)
Polarization in cartesian coordinates (a.u.):
Total: -0.282749182E-01 -0.282749182E-01 -0.282749182E-01
Polarization in cartesian coordinates (a.u.):
Total: -0.282310128E-01 -0.282310128E-01 -0.282310128E-01
Polarization in cartesian coordinates (a.u.):
Total: -0.283187730E-01 -0.283187730E-01 -0.283187730E-01
In a finite electric field, the polarization in terms of the linear and quadratic susceptibilities is, in SI units,
or, in atomic units:
as \(4\pi\epsilon_0 = 1\) in atomic units.
The change of polarization for positive and negative fields above are nearly the same, showing again that the quadratic term is almost negligible. This clearly appears in the figure below where the field dependence of the polarization for a larger range of electric fields is plotted.
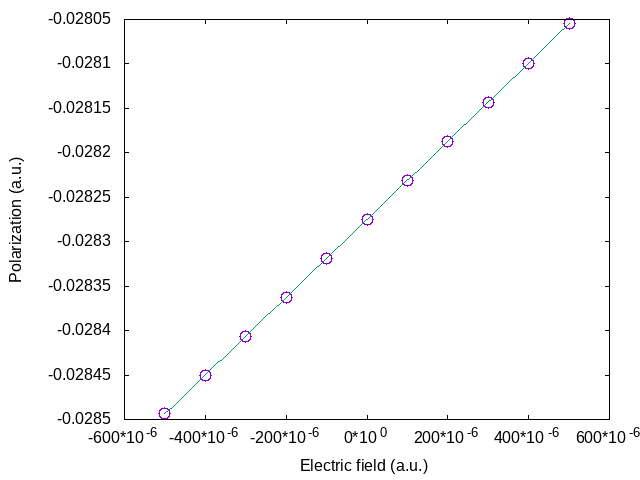
We can therefore extract the linear optical dielectric susceptibility:
The relative permittivity, also known as the dielectric constant, is
This value is a bit over the value of 6.463 obtained by DFPT from tpolarization_3.abi. Typically, finite field calculations converge with the density of the k point grid more slowly than DFPT calculations.
If you do calculations for more electric fields, fitting them with the general
expression of the polarization above (including the \(E^2\) term) you can find the non-
linear optical susceptibility \(\chi^{(2)}/4\pi\) (in atomic units).
For tpolarization_6.abi you should find \(\chi^{(2)}/4\pi = 2.5427\), so
in SI units \(\chi^{(2)} = 62.14~\mathrm{pm/V}\) and \(d_{36} = 15.54~\mathrm{pm/V}\).
The relationship between the \(\chi^{(2)}_{ijk}\) tensor and the \(d_{ij}\) tensor (the
quantity reported by abinit in a nonlinear optics DFPT computation) involves
a variety of symmetries and is explained in detail in the book [Boyd2020].
Looking at the evolution of the stress with electric field (see graphic below), you should also be able to extract the piezoelectric constants. You can try to do it as an exercise. As the calculation here was at clamped ions, you will get the clamped ion proper piezoelectric constants. You should obtain -0.6365 C/m\(^2\). The relationships between the various response functions under conditions of strain, stress, field, and so forth are discussed in depth in [Wu2005].
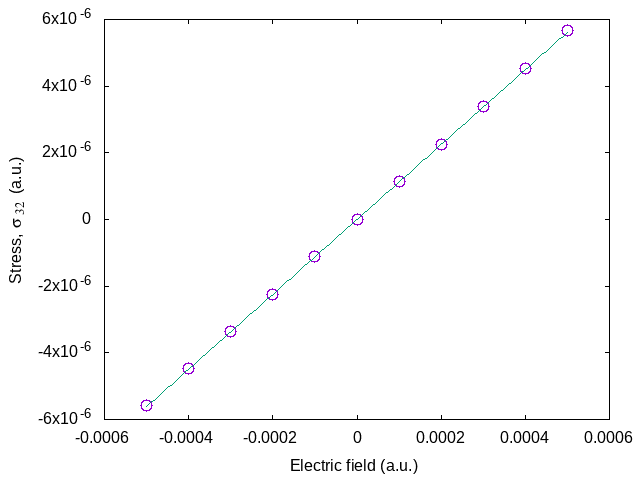
You can modify the previous input file to perform a finite field calculation combined with ion relaxation, similarly to how tpolarization_5.abi was modified from tpolarization_4.abi, giving access to the the relaxed ion proper piezoelectric constant. From the output of this run, analyzing the results in the same way as before, you should obtain 0.1311 C/m\(^2\) for the relaxed ion piezoelectric constant.