How to build ABINIT using the Autotools¶
This tutorial explains how to build ABINIT including the external dependencies without relying on pre-compiled libraries, package managers and root privileges. You will learn how to use the standard configure and make Linux tools to build and install your own software stack including the MPI library and the associated mpif90 and mpicc wrappers required to compile MPI applications.
It is assumed that you already have a standard Unix-like installation that provides the basic tools needed to build software from source (Fortran/C compilers and make). The changes required for macOS are briefly mentioned when needed. Windows users should install cygwin that provides a POSIX-compatible environment or, alternatively, use a Windows Subsystem for Linux. Note that the procedure described in this tutorial has been tested with Linux/macOS hence feedback and suggestions from Windows users are welcome.
Tip
In the last part of the tutorial, we discuss more advanced topics such as using modules in supercomputing centers, compiling and linking with the intel compilers and the MKL library as well as OpenMP threads. You may want to jump directly to this section if you are already familiar with software compilation.
In the following, we will make extensive use of the bash shell hence familiarity with the terminal is assumed. For a quick introduction to the command line, please consult this Ubuntu tutorial. If this is the first time you use the configure && make approach to build software, we strongly recommend to read this guide before proceeding with the next steps.
If, on the other hand, you are not interested in compiling all the components from source, you may want to consider the following alternatives:
-
Compilation with external libraries provided by apt-based Linux distributions (e.g. Ubuntu). More info available here.
-
Compilation with external libraries on Fedora/RHEL/CentOS Linux distributions. More info available here.
-
Homebrew bottles or macports for macOS. More info available here.
-
Automatic compilation and generation of modules on clusters with EasyBuild. More info available here.
-
Compiling Abinit using the internal fallbacks and the build-abinit-fallbacks.sh script automatically generated by configure if the mandatory dependencies are not found.
-
Using precompiled binaries provided by conda-forge (for Linux and macOS users).
Before starting, it is also worth reading this document prepared by Marc Torrent that introduces important concepts and provides a detailed description of the configuration options supported by the ABINIT build system. Note that these slides have been written for Abinit v8 hence some examples should be changed in order to be compatible with the build system of version 9, yet the document represents a valuable source of information.
Important
The aim of this tutorial is to teach you how to compile code from source but we cannot guarantee that these recipes will work out of the box on every possible architecture. We will do our best to explain how to setup your environment and how to avoid the typical pitfalls but we cannot cover all the possible cases.
Fortunately, the internet provides lots of resources. Search engines and stackoverflow are your best friends and in some cases one can find the solution by just copying the error message in the search bar. For more complicated issues, you can ask for help on the ABINIT discourse forum or contact the sysadmin of your cluster but remember to provide enough information about your system and the problem you are encountering.
Getting started¶
Since ABINIT is written in Fortran, we need a recent Fortran compiler that supports the F2003 specifications as well as a C compiler. At the time of writing (May 09, 2026 ), the C++ compiler is optional and required only for advanced features that are not treated in this tutorial.
In what follows, we will be focusing on the GNU toolchain i.e. gcc for C and gfortran for Fortran. These “sequential” compilers are adequate if you don’t need to compile parallel MPI applications. The compilation of MPI code, indeed, requires the installation of additional libraries and specialized wrappers (mpicc, mpif90 or mpiifort ) replacing the “sequential” compilers. This very important scenario is covered in more detail in the next sections. For the time being, we mainly focus on the compilation of sequential applications/libraries.
First of all, let’s make sure the gfortran compiler is installed on your machine by issuing in the terminal the following command:
which gfortran
/usr/bin/gfortran
Tip
The which command, returns the absolute path of the executable. This Unix tool is extremely useful to pinpoint possible problems and we will use it a lot in the rest of this tutorial.
In our case, we are lucky that the Fortran compiler is already installed in /usr/bin and we can immediately use it to build our software stack. If gfortran is not installed, you may want to use the package manager provided by your Linux distribution to install it. On Ubuntu, for instance, use:
sudo apt-get install gfortran
To get the version of the compiler, use the --version option:
gfortran --version
GNU Fortran (GCC) 5.3.1 20160406 (Red Hat 5.3.1-6)
Copyright (C) 2015 Free Software Foundation, Inc.
Starting with version 9, ABINIT requires gfortran >= v5.4. Consult the release notes to check whether your gfortran version is supported by the latest ABINIT releases.
Now let’s check whether make is already installed using:
which make
/usr/bin/make
Hopefully, the C compiler gcc is already installed on your machine.
which gcc
/usr/bin/gcc
At this point, we have all the basic building blocks needed to compile ABINIT from source and we can proceed with the next steps.
Tip
Life gets hard if you are a macOS user as Apple does not officially support Fortran (😞) so you need to install gfortran and gcc either via homebrew or macport. Alternatively, one can install gfortran using one of the standalone DMG installers provided by the gfortran-for-macOS project. Note also that macOS users will need to install make via Xcode. More info can be found in this page.
How to compile BLAS and LAPACK¶
BLAS and LAPACK represent the workhorse of many scientific codes and an optimized implementation is crucial for achieving good performance. In principle this step can be skipped as any decent Linux distribution already provides pre-compiled versions but, as already mentioned in the introduction, we are geeks and we prefer to compile everything from source. Moreover the compilation of BLAS/LAPACK represents an excellent exercise that gives us the opportunity to discuss some basic concepts that will reveal very useful in the other parts of this tutorial.
First of all, let’s create a new directory inside your $HOME (let’s call it local) using the command:
cd $HOME && mkdir local
Tip
$HOME is a standard shell variable that stores the absolute path to your home directory. Use:
echo My home directory is $HOME
to print the value of the variable.
The && syntax is used to chain commands together, such that the next command is executed if and only if the preceding command exited without errors (or, more accurately, exits with a return code of 0). We will use this trick a lot in the other examples to reduce the number of lines we have to type in the terminal so that one can easily cut and paste the examples in the terminal.
Now create the src subdirectory inside $HOME/local with:
cd $HOME/local && mkdir src && cd src
The src directory will be used to store the packages with the source files and compile code,
whereas executables and libraries will be installed in $HOME/local/bin and $HOME/local/lib, respectively.
We use $HOME/local because we are working as normal users and we cannot install software
in /usr/local where root privileges are required and a sudo make install would be needed.
Moreover, working inside $HOME/local allows us to keep our software stack well separated
from the libraries installed by our Linux distribution so that we can easily test new libraries and/or
different versions without affecting the software stack installed by our distribution.
Now download the tarball from the openblas website with:
wget https://github.com/xianyi/OpenBLAS/archive/v0.3.7.tar.gz
If wget is not available, use curl with the -o option to specify the name of the output file as in:
curl -L https://github.com/xianyi/OpenBLAS/archive/v0.3.7.tar.gz -o v0.3.7.tar.gz
Tip
To get the URL associated to a HTML link inside the browser, hover the mouse pointer over the link,
press the right mouse button and then select Copy Link Address to copy the link to the system clipboard.
Then paste the text in the terminal by selecting the Copy action in the menu
activated by clicking on the right button.
Alternatively, one can press the central button (mouse wheel) or use CMD + V on macOS.
This trick is quite handy to fetch tarballs directly from the terminal.
Uncompress the tarball with:
tar -xvf v0.3.7.tar.gz
then cd to the directory with:
cd OpenBLAS-0.3.7
and execute
make -j2 USE_THREAD=0 USE_LOCKING=1
to build the single thread version.
Tip
By default, openblas activates threads (see FAQ page)
but in our case we prefer to use the sequential version as Abinit is mainly optimized for MPI.
The -j2 option tells make to use 2 processes to build the code in order to speed up the compilation.
Adjust this value according to the number of physical cores available on your machine.
At the end of the compilation, you should get the following output (note Single threaded):
OpenBLAS build complete. (BLAS CBLAS LAPACK LAPACKE)
OS ... Linux
Architecture ... x86_64
BINARY ... 64bit
C compiler ... GCC (command line : cc)
Fortran compiler ... GFORTRAN (command line : gfortran)
Library Name ... libopenblas_haswell-r0.3.7.a (Single threaded)
To install the library, you can run "make PREFIX=/path/to/your/installation install".
You may have noticed that, in this particular case, make is not just building the library but is also running unit tests to validate the build. This means that if make completes successfully, we can be confident that the build is OK and we can proceed with the installation. Other packages use a more standard approach and provide a make check option that should be executed after make in order to run the test suite before installing the package.
To install openblas in $HOME/local, issue:
make PREFIX=$HOME/local/ install
At this point, we should have the following include files installed in $HOME/local/include:
ls $HOME/local/include/
cblas.h f77blas.h lapacke.h lapacke_config.h lapacke_mangling.h lapacke_utils.h openblas_config.h
and the following libraries installed in $HOME/local/lib:
ls $HOME/local/lib/libopenblas*
/home/gmatteo/local/lib/libopenblas.a /home/gmatteo/local/lib/libopenblas_haswell-r0.3.7.a
/home/gmatteo/local/lib/libopenblas.so /home/gmatteo/local/lib/libopenblas_haswell-r0.3.7.so
/home/gmatteo/local/lib/libopenblas.so.0
Files ending with .so are shared libraries (.so stands for shared object) whereas
.a files are static libraries.
When compiling source code that relies on external libraries, the name of the library
(without the lib prefix and the file extension) as well as the directory where the library is located must be passed
to the linker.
The name of the library is usually specified with the -l option while the directory is given by -L.
According to these simple rules, in order to compile source code that uses BLAS/LAPACK routines,
one should use the following option:
-L$HOME/local/lib -lopenblas
We will use a similar syntax to help the ABINIT configure script locate the external linear algebra library.
Important
You may have noticed that we haven’t specified the file extension in the library name.
If both static and shared libraries are found, the linker gives preference to linking with the shared library
unless the -static option is used.
Dynamic is the default behaviour on several Linux distributions so we assume dynamic linking
in what follows.
If you are compiling C or Fortran code that requires include files with the declaration of prototypes and the definition
of named constants, you will need to specify the location of the include files via the -I option.
In this case, the previous options should be augmented by:
-L$HOME/local/lib -lopenblas -I$HOME/local/include
This approach is quite common for C code where .h files must be included to compile properly.
It is less common for modern Fortran code in which include files are usually replaced by .mod files
i.e. Fortran modules produced by the compiler whose location is usually specified via the -J option.
Still, the -I option for include files is valuable also when compiling Fortran applications as libraries
such as FFTW and MKL rely on (Fortran) include files whose location should be passed to the compiler
via -I instead of -J,
see also the official gfortran documentation.
Do not worry if this rather technical point is not clear to you.
Any external library has its own requirements and peculiarities and the ABINIT build system provides several options
to automate the detection of external dependencies and the final linkage.
The most important thing is that you are now aware that the compilation of ABINIT requires
the correct specification of -L, -l for libraries, -I for include files, and -J for Fortran modules.
We will elaborate more on this topic when we discuss the configuration options supported by the ABINIT build system.
Since we have installed the package in a non-standard directory ($HOME/local),
we need to update two important shell variables: $PATH and $LD_LIBRARY_PATH.
If this is the first time you hear about $PATH and $LD_LIBRARY_PATH, please take some time to learn
about the meaning of these environment variables.
More information about $PATH is available here.
See this page for $LD_LIBRARY_PATH.
Add these two lines at the end of your $HOME/.bash_profile file
export PATH=$HOME/local/bin:$PATH
export LD_LIBRARY_PATH=$HOME/local/lib:$LD_LIBRARY_PATH
then execute:
source $HOME/.bash_profile
to activate these changes without having to start a new terminal session. Now use:
echo $PATH
echo $LD_LIBRARY_PATH
to print the value of these variables. On my Linux box, I get:
echo $PATH
/home/gmatteo/local/bin:/usr/local/bin:/usr/bin:/usr/local/sbin:/usr/sbin
echo $LD_LIBRARY_PATH
/home/gmatteo/local/lib:
Note how /home/gmatteo/local/bin has been prepended to the previous value of $PATH.
From now on, we can invoke any executable located in $HOME/local/bin by just typing
its base name in the shell without having to the enter the full path.
Warning
Using:
export PATH=$HOME/local/bin
is not a very good idea as the shell will stop working. Can you explain why?
Tip
macOS users should replace LD_LIBRARY_PATH with DYLD_LIBRARY_PATH
Remember also that one can use env to print all the environment variables defined
in your session and pipe the results to other Unix tools.
Try e.g.:
env | grep LD_
to print only the variables whose name starts with LD_
We conclude this section with another tip. From time to time, some compilers complain or do not display important messages because language support is improperly configured on your computer. Should this happen, we recommend to export the two variables:
export LANG=C
export LC_ALL=C
This will reset the language support to its most basic defaults and will make sure that you get all messages from the compilers in English.
How to compile libxc¶
At this point, it should not be so difficult to compile and install
libxc, a library that provides
many useful XC functionals (PBE, meta-GGA, hybrid functionals, etc).
Libxc is written in C and can be built using the standard configure && make approach.
No external dependency is needed, except for basic C libraries that are available
on any decent Linux distribution.
Let’s start by fetching the tarball from the internet:
# Get the tarball.
# Note the -O option used in wget to specify the name of the output file
cd $HOME/local/src
wget http://www.tddft.org/programs/libxc/down.php?file=4.3.4/libxc-4.3.4.tar.gz -O libxc.tar.gz
tar -zxvf libxc.tar.gz
Now configure the package with the standard --prefix option
to specify the location where all the libraries, executables, include files,
Fortran modules, man pages, etc. will be installed when we execute make install
(the default is /usr/local)
cd libxc-4.3.4 && ./configure --prefix=$HOME/local
Finally, build the library, run the tests and install it with:
make -j2
make check && make install
At this point, we should have the following include files in $HOME/local/include
[gmatteo@bob libxc-4.3.4]$ ls ~/local/include/*xc*
/home/gmatteo/local/include/libxc_funcs_m.mod /home/gmatteo/local/include/xc_f90_types_m.mod
/home/gmatteo/local/include/xc.h /home/gmatteo/local/include/xc_funcs.h
/home/gmatteo/local/include/xc_f03_lib_m.mod /home/gmatteo/local/include/xc_funcs_removed.h
/home/gmatteo/local/include/xc_f90_lib_m.mod /home/gmatteo/local/include/xc_version.h
where .mod are Fortran modules generated by the compiler that are needed
when compiling Fortran source using the libxc Fortran API.
Warning
The .mod files are compiler- and version-dependent.
In other words, one cannot use these .mod files to compile code with a different Fortran compiler.
Moreover, you should not expect to be able to use modules compiled with
a different version of the same compiler, especially if the major version has changed.
This is one of the reasons why the version of the Fortran compiler employed
to build our software stack is very important.
Finally, we have the following static libraries installed in ~/local/lib
ls ~/local/lib/libxc*
/home/gmatteo/local/lib/libxc.a /home/gmatteo/local/lib/libxcf03.a /home/gmatteo/local/lib/libxcf90.a
/home/gmatteo/local/lib/libxc.la /home/gmatteo/local/lib/libxcf03.la /home/gmatteo/local/lib/libxcf90.la
where:
- libxc is the C library
- libxcf90 is the library with the F90 API
- libxcf03 is the library with the F2003 API
Both libxcf90 and libxcf03 depend on the C library where most of the work is done. At present, ABINIT requires the F90 API only so we should use
-L$HOME/local/lib -lxcf90 -lxc
for the libraries and
-I$HOME/local/include
for the include files.
Note how libxcf90 comes before the C library libxc.
This is done on purpose as libxcf90 depends on libxc (the Fortran API calls the C implementation).
Inverting the order of the libraries will likely trigger errors (undefined references)
in the last step of the compilation when the linker tries to build the final application.
Things become even more complicated when we have to build applications using many different interdependent libraries as the order of the libraries passed to the linker is of crucial importance. Fortunately the ABINIT build system is aware of this problem and all the dependencies (BLAS, LAPACK, FFT, LIBXC, MPI, etc) will be automatically put in the right order so you don’t have to worry about this point although it is worth knowing about it.
Compiling and installing FFTW¶
FFTW is a C library for computing the Fast Fourier transform in one or more dimensions. ABINIT already provides an internal implementation of the FFT algorithm implemented in Fortran hence FFTW is considered an optional dependency. Nevertheless, we do not recommend the internal implementation if you really care about performance. The reason is that FFTW (or, even better, the DFTI library provided by intel MKL) is usually much faster than the internal version.
Important
FFTW is very easy to install on Linux machines once you have gcc and gfortran. The fftalg variable defines the implementation to be used and 312 corresponds to the FFTW implementation. The default value of fftalg is automatically set by the configure script via pre-preprocessing options. In other words, if you activate support for FFTW (DFTI) at configure time, ABINIT will use fftalg 312 (512) as default.
The FFTW source code can be downloaded from fftw.org, and the tarball of the latest version is available at http://www.fftw.org/fftw-3.3.8.tar.gz.
cd $HOME/local/src
wget http://www.fftw.org/fftw-3.3.8.tar.gz
tar -zxvf fftw-3.3.8.tar.gz && cd fftw-3.3.8
The compilation procedure is very similar to the one already used for the libxc package. Note, however, that ABINIT needs both the single-precision and the double-precision version. This means that we need to configure, build and install the package twice.
To build the single precision version, use:
./configure --prefix=$HOME/local --enable-single
make -j2
make check && make install
During the configuration step, make sure that configure finds the Fortran compiler because ABINIT needs the Fortran interface.
checking for gfortran... gfortran
checking whether we are using the GNU Fortran 77 compiler... yes
checking whether gfortran accepts -g... yes
checking if libtool supports shared libraries... yes
checking whether to build shared libraries... no
checking whether to build static libraries... yes
Let’s have a look at the libraries we’ve just installed:
ls $HOME/local/lib/libfftw3*
/home/gmatteo/local/lib/libfftw3f.a /home/gmatteo/local/lib/libfftw3f.la
the f at the end stands for float (C jargon for single precision).
Note that only static libraries have been built.
To build shared libraries, one should use --enable-shared when configuring.
Now we configure for the double precision version (this is the default behaviour so no extra option is needed)
./configure --prefix=$HOME/local
make -j2
make check && make install
After this step, you should have two libraries with the single and the double precision API:
ls $HOME/local/lib/libfftw3*
/home/gmatteo/local/lib/libfftw3.a /home/gmatteo/local/lib/libfftw3f.a
/home/gmatteo/local/lib/libfftw3.la /home/gmatteo/local/lib/libfftw3f.la
To compile ABINIT with FFTW3 support, one should use:
-L$HOME/local/lib -lfftw3f -lfftw3 -I$HOME/local/include
Note that, unlike in libxc, here we don’t have to specify different libraries for Fortran and C as FFTW3 bundles both the C and the Fortran API in the same library. The Fortran interface is included by default provided the FFTW3 configure script can find a Fortran compiler. In our case, we know that our FFTW3 library supports Fortran as gfortran was found by configure but this may not be true if you are using a precompiled library installed via your package manager.
To make sure we have the Fortran API, use the nm tool
to get the list of symbols in the library and then use grep to search for the Fortran API.
For instance we can check whether our library contains the Fortran routine for multiple single-precision
FFTs (sfftw_plan_many_dft) and the version for multiple double-precision FFTs (dfftw_plan_many_dft)
[gmatteo@bob fftw-3.3.8]$ nm $HOME/local/lib/libfftw3f.a | grep sfftw_plan_many_dft
0000000000000400 T sfftw_plan_many_dft_
0000000000003570 T sfftw_plan_many_dft__
0000000000001a90 T sfftw_plan_many_dft_c2r_
0000000000004c00 T sfftw_plan_many_dft_c2r__
0000000000000f60 T sfftw_plan_many_dft_r2c_
00000000000040d0 T sfftw_plan_many_dft_r2c__
[gmatteo@bob fftw-3.3.8]$ nm $HOME/local/lib/libfftw3.a | grep dfftw_plan_many_dft
0000000000000400 T dfftw_plan_many_dft_
0000000000003570 T dfftw_plan_many_dft__
0000000000001a90 T dfftw_plan_many_dft_c2r_
0000000000004c00 T dfftw_plan_many_dft_c2r__
0000000000000f60 T dfftw_plan_many_dft_r2c_
00000000000040d0 T dfftw_plan_many_dft_r2c__
If you are using a FFTW3 library without Fortran support, the ABINIT configure script will complain that the library cannot be called from Fortran and you will need to dig into config.log to understand what’s going on.
Note
At present, there is no need to compile FFTW with MPI support because ABINIT implements its own version of the MPI-FFT algorithm based on the sequential FFTW version. The MPI algorithm implemented in ABINIT is optimized for plane-waves codes as it supports zero-padding and composite transforms for the applications of the local part of the KS potential.
Also, do not use MKL with FFTW3 for the FFT as the MKL library exports the same symbols as FFTW. This means that the linker will receive multiple definitions for the same procedure and the behaviour is undefined! Use either MKL or FFTW3 with e.g. openblas.
Installing MPI¶
In this section, we discuss how to compile and install the MPI library. This step is required if you want to run ABINIT with multiple processes and/or you need to compile MPI-based libraries such as PBLAS/Scalapack or the HDF5 library with support for parallel IO.
It is worth stressing that the MPI installation provides two scripts (mpif90 and mpicc) that act as a sort of wrapper around the sequential Fortran and the C compilers, respectively. These scripts must be used to compile parallel software using MPI instead of the “sequential” gfortran and gcc. The MPI library also provides launcher scripts installed in the bin directory (mpirun or mpiexec) that must be used to execute an MPI application EXEC with NUM_PROCS MPI processes with the syntax:
mpirun -n NUM_PROCS EXEC [EXEC_ARGS]
Warning
Keep in mind that there are several MPI implementations available around (openmpi, mpich, intel mpi, etc) and you must choose one implementation and stick to it when building your software stack. In other words, all the libraries and executables requiring MPI must be compiled, linked and executed with the same MPI library.
Don’t try to link a library compiled with e.g. mpich if you are building the code with the mpif90 wrapper provided by e.g. openmpi. By the same token, don’t try to run executables compiled with e.g. intel mpi with the mpirun launcher provided by openmpi unless you are looking for troubles! Again, the which command is quite handy to pinpoint possible problems especially if there are multiple installations of MPI in your $PATH (not a very good idea!).
In this tutorial, we employ the mpich implementation that can be downloaded from this webpage. In the terminal, issue:
cd $HOME/local/src
wget http://www.mpich.org/static/downloads/3.3.2/mpich-3.3.2.tar.gz
tar -zxvf mpich-3.3.2.tar.gz
cd mpich-3.3.2/
to download and uncompress the tarball. Then configure/compile/test/install the library with:
./configure --prefix=$HOME/local
make -j2
make check && make install
Once the installation is completed, you should obtain this message (possibly not the last message, you might have to look for it).
----------------------------------------------------------------------
Libraries have been installed in:
/home/gmatteo/local/lib
If you ever happen to want to link against installed libraries
in a given directory, LIBDIR, you must either use libtool, and
specify the full pathname of the library, or use the '-LLIBDIR'
flag during linking and do at least one of the following:
- add LIBDIR to the 'LD_LIBRARY_PATH' environment variable
during execution
- add LIBDIR to the 'LD_RUN_PATH' environment variable
during linking
- use the '-Wl,-rpath -Wl,LIBDIR' linker flag
- have your system administrator add LIBDIR to '/etc/ld.so.conf'
See any operating system documentation about shared libraries for
more information, such as the ld(1) and ld.so(8) manual pages.
----------------------------------------------------------------------
The reason why we should add $HOME/local/lib to $LD_LIBRARY_PATH now should be clear to you.
Let’s have a look at the MPI executables we have just installed in $HOME/local/bin:
ls $HOME/local/bin/mpi*
/home/gmatteo/local/bin/mpic++ /home/gmatteo/local/bin/mpiexec /home/gmatteo/local/bin/mpifort
/home/gmatteo/local/bin/mpicc /home/gmatteo/local/bin/mpiexec.hydra /home/gmatteo/local/bin/mpirun
/home/gmatteo/local/bin/mpichversion /home/gmatteo/local/bin/mpif77 /home/gmatteo/local/bin/mpivars
/home/gmatteo/local/bin/mpicxx /home/gmatteo/local/bin/mpif90
Since we added $HOME/local/bin to $PATH, we should see that mpi90 is actually pointing to the version we have just installed:
which mpif90
~/local/bin/mpif90
As already mentioned, mpif90 is a wrapper around the sequential Fortran compiler. To show the Fortran compiler invoked by mpif90, use:
mpif90 -v
mpifort for MPICH version 3.3.2
Using built-in specs.
COLLECT_GCC=gfortran
COLLECT_LTO_WRAPPER=/usr/libexec/gcc/x86_64-redhat-linux/5.3.1/lto-wrapper
Target: x86_64-redhat-linux
Configured with: ../configure --enable-bootstrap --enable-languages=c,c++,objc,obj-c++,fortran,ada,go,lto --prefix=/usr --mandir=/usr/share/man --infodir=/usr/share/info --with-bugurl=http://bugzilla.redhat.com/bugzilla --enable-shared --enable-threads=posix --enable-checking=release --enable-multilib --with-system-zlib --enable-__cxa_atexit --disable-libunwind-exceptions --enable-gnu-unique-object --enable-linker-build-id --with-linker-hash-style=gnu --enable-plugin --enable-initfini-array --disable-libgcj --with-isl --enable-libmpx --enable-gnu-indirect-function --with-tune=generic --with-arch_32=i686 --build=x86_64-redhat-linux
Thread model: posix
gcc version 5.3.1 20160406 (Red Hat 5.3.1-6) (GCC)
The C include files (.h) and the Fortran modules (.mod) have been installed in $HOME/local/include
ls $HOME/local/include/mpi*
/home/gmatteo/local/include/mpi.h /home/gmatteo/local/include/mpicxx.h
/home/gmatteo/local/include/mpi.mod /home/gmatteo/local/include/mpif.h
/home/gmatteo/local/include/mpi_base.mod /home/gmatteo/local/include/mpio.h
/home/gmatteo/local/include/mpi_constants.mod /home/gmatteo/local/include/mpiof.h
/home/gmatteo/local/include/mpi_sizeofs.mod
In principle, the location of the directory must be passed to the Fortran compiler either
with the -J (mpi.mod module for MPI2+) or the -I option (mpif.h include file for MPI1).
Fortunately, the ABINIT build system can automatically detect your MPI installation and set all the compilation
options automatically if you provide the installation root ($HOME/local).
Installing HDF5 and netcdf4¶
Abinit developers are trying to move away from Fortran binary files as this format is not portable and difficult to read from high-level languages such as python. For this reason, in Abinit v9, HDF5 and netcdf4 have become hard-requirements. This means that the configure script will abort if these libraries are not found. In this section, we explain how to build HDF5 and netcdf4 from source including support for parallel IO.
Netcdf4 is built on top of HDF5 and consists of two different layers:
-
The low-level C library
-
The Fortran bindings i.e. Fortran routines calling the low-level C implementation. This is the high-level API used by ABINIT to perform all the IO operations on netcdf files.
To build the libraries required by ABINIT, we will compile the three different layers in a bottom-up fashion starting from the HDF5 package (HDF5 → netcdf-c → netcdf-fortran). Since we want to activate support for parallel IO, we need to compile the libraries using the wrappers provided by our MPI installation instead of using gcc or gfortran directly.
Let’s start by downloading the HDF5 tarball from this download page. Uncompress the archive with tar as usual, then configure the package with:
./configure --prefix=$HOME/local/ \
CC=$HOME/local/bin/mpicc --enable-parallel --enable-shared
where we’ve used the CC variable to specify the C compiler. This step is crucial in order to activate support for parallel IO.
Tip
A table with the more commonly-used predefined variables is available here
At the end of the configuration step, you should get the following output:
AM C Flags:
Shared C Library: yes
Static C Library: yes
Fortran: no
C++: no
Java: no
Features:
---------
Parallel HDF5: yes
Parallel Filtered Dataset Writes: yes
Large Parallel I/O: yes
High-level library: yes
Threadsafety: no
Default API mapping: v110
With deprecated public symbols: yes
I/O filters (external): deflate(zlib)
MPE:
Direct VFD: no
dmalloc: no
Packages w/ extra debug output: none
API tracing: no
Using memory checker: no
Memory allocation sanity checks: no
Function stack tracing: no
Strict file format checks: no
Optimization instrumentation: no
The line with:
Parallel HDF5: yes
tells us that our HDF5 build supports parallel IO. The Fortran API is not activated but this is not a problem as ABINIT will be interfaced with HDF5 through the Fortran bindings provided by netcdf-fortran. In other words, ABINIT requires netcdf-fortran and not the HDF5 Fortran bindings.
Again, issue make -j NUM followed by make check and finally make install.
Note that make check may take some time so you may want to install immediately and run the tests in another terminal
so that you can continue with the tutorial.
Now let’s move to netcdf. Download the C version and the Fortran bindings from the netcdf website and unpack the tarball files as usual.
wget ftp://ftp.unidata.ucar.edu/pub/netcdf/netcdf-c-4.7.3.tar.gz
tar -xvf netcdf-c-4.7.3.tar.gz
wget ftp://ftp.unidata.ucar.edu/pub/netcdf/netcdf-fortran-4.5.2.tar.gz
tar -xvf netcdf-fortran-4.5.2.tar.gz
To compile the C library, use:
cd netcdf-c-4.7.3
./configure --prefix=$HOME/local/ \
CC=$HOME/local/bin/mpicc \
LDFLAGS=-L$HOME/local/lib CPPFLAGS=-I$HOME/local/include
where mpicc is used as C compiler (CC environment variable)
and we have to specify LDFLAGS and CPPFLAGS as we want to link against our installation of hdf5.
At the end of the configuration step, we should obtain
# NetCDF C Configuration Summary
==============================
# General
-------
NetCDF Version: 4.7.3
Dispatch Version: 1
Configured On: Wed Apr 8 00:53:19 CEST 2020
Host System: x86_64-pc-linux-gnu
Build Directory: /home/gmatteo/local/src/netcdf-c-4.7.3
Install Prefix: /home/gmatteo/local
# Compiling Options
-----------------
C Compiler: /home/gmatteo/local/bin/mpicc
CFLAGS:
CPPFLAGS: -I/home/gmatteo/local/include
LDFLAGS: -L/home/gmatteo/local/lib
AM_CFLAGS:
AM_CPPFLAGS:
AM_LDFLAGS:
Shared Library: yes
Static Library: yes
Extra libraries: -lhdf5_hl -lhdf5 -lm -ldl -lz -lcurl
# Features
--------
NetCDF-2 API: yes
HDF4 Support: no
HDF5 Support: yes
NetCDF-4 API: yes
NC-4 Parallel Support: yes
PnetCDF Support: no
DAP2 Support: yes
DAP4 Support: yes
Byte-Range Support: no
Diskless Support: yes
MMap Support: no
JNA Support: no
CDF5 Support: yes
ERANGE Fill Support: no
Relaxed Boundary Check: yes
The section:
HDF5 Support: yes
NetCDF-4 API: yes
NC-4 Parallel Support: yes
tells us that configure detected our installation of hdf5 and that support for parallel-IO is activated.
Now use the standard sequence of commands to compile and install the package:
make -j2
make check && make install
Once the installation is completed, use the nc-config executable to
inspect the features provided by the library we’ve just installed.
which nc-config
/home/gmatteo/local/bin/nc-config
# installation directory
nc-config --prefix
/home/gmatteo/local/
To get a summary of the options used to build the C layer and the available features, use
nc-config --all
This netCDF 4.7.3 has been built with the following features:
--cc -> /home/gmatteo/local/bin/mpicc
--cflags -> -I/home/gmatteo/local/include
--libs -> -L/home/gmatteo/local/lib -lnetcdf
--static -> -lhdf5_hl -lhdf5 -lm -ldl -lz -lcurl
....
<snip>
nc-config is quite useful as it prints the compiler options required to
build C applications requiring netcdf-c (--cflags and --libs).
Unfortunately, this tool is not enough for ABINIT as we need the Fortran bindings as well.
To compile the Fortran bindings, execute:
cd netcdf-fortran-4.5.2
./configure --prefix=$HOME/local/ \
FC=$HOME/local/bin/mpif90 \
LDFLAGS=-L$HOME/local/lib FCFLAGS=-I$HOME/local/include
where FC points to our mpif90 wrapper (CC is not needed here). For further info on how to build netcdf-fortran, see the official documentation.
Now issue:
make -j2
make check && make install
To inspect the features activated in our Fortran library, use nf-config instead of nc-config
(note the nf- prefix):
which nf-config
/home/gmatteo/local/bin/nf-config
# installation directory
nf-config --prefix
/home/gmatteo/local/
To get a summary of the options used to build the Fortran bindings and the list of available features, use
nf-config --all
This netCDF-Fortran 4.5.2 has been built with the following features:
--cc -> gcc
--cflags -> -I/home/gmatteo/local/include -I/home/gmatteo/local/include
--fc -> /home/gmatteo/local/bin/mpif90
--fflags -> -I/home/gmatteo/local/include
--flibs -> -L/home/gmatteo/local/lib -lnetcdff -L/home/gmatteo/local/lib -lnetcdf -lnetcdf -ldl -lm
--has-f90 ->
--has-f03 -> yes
--has-nc2 -> yes
--has-nc4 -> yes
--prefix -> /home/gmatteo/local
--includedir-> /home/gmatteo/local/include
--version -> netCDF-Fortran 4.5.2
Tip
nf-config is quite handy to pass options to the ABINIT configure script.
Instead of typing the full list of libraries (--flibs) and the location of the include files (--fflags)
we can delegate this boring task to nf-config using
backtick syntax:
NETCDF_FORTRAN_LIBS=`nf-config --flibs`
NETCDF_FORTRAN_FCFLAGS=`nf-config --fflags`
Alternatively, one can simply pass the installation directory (here we use the $(...) syntax):
--with-netcdf-fortran=$(nf-config --prefix)
and then let configure detect NETCDF_FORTRAN_LIBS and NETCDF_FORTRAN_FCFLAGS for us.
How to compile ABINIT¶
In this section, we finally discuss how to compile ABINIT using the MPI compilers and the libraries installed previously. First of all, download the ABINIT tarball from this page using e.g.
wget https://www.abinit.org/sites/default/files/packages/abinit-9.10.3.tar.gz
Here we are using version 9.10.3 but you may want to download the latest production version to take advantage of new features and benefit from bug fixes.
Once you got the tarball, uncompress it by typing:
tar -xvzf abinit-9.10.3.tar.gz
Then cd into the newly created abinit-9.10.3 directory.
Before actually starting the compilation, type:
./configure --help
and take some time to read the documentation of the different options.
The documentation mentions the most important environment variables that can be used to specify compilers and compilation flags. We already encountered some of these variables in the previous examples:
Some influential environment variables:
CC C compiler command
CFLAGS C compiler flags
LDFLAGS linker flags, e.g. -L<lib dir> if you have libraries in a
nonstandard directory <lib dir>
LIBS libraries to pass to the linker, e.g. -l<library>
CPPFLAGS (Objective) C/C++ preprocessor flags, e.g. -I<include dir> if
you have headers in a nonstandard directory <include dir>
CPP C preprocessor
CXX C++ compiler command
CXXFLAGS C++ compiler flags
FC Fortran compiler command
FCFLAGS Fortran compiler flags
Besides the standard environment variables: CC, CFLAGS, FC, FCFLAGS etc. the build system also provides specialized options to activate support for external libraries. For libxc, for instance, we have:
LIBXC_CPPFLAGS
C preprocessing flags for LibXC.
LIBXC_CFLAGS
C flags for LibXC.
LIBXC_FCFLAGS
Fortran flags for LibXC.
LIBXC_LDFLAGS
Linker flags for LibXC.
LIBXC_LIBS
Library flags for LibXC.
According to what we have seen during the compilation of libxc, one should pass to configure the following options:
LIBXC_LIBS="-L$HOME/local/lib -lxcf90 -lxc"
LIBXC_FCFLAGS="-I$HOME/local/include"
Alternatively, one can use the high-level interface provided by the --with-LIBNAME options
to specify the installation directory as in:
--with-libxc="$HOME/local/lib"
In this case, configure will try to automatically detect the other options.
This is the easiest approach but if configure cannot detect the dependency properly,
you may need to inspect config.log for error messages and/or set the options manually.
In the previous examples, we executed configure in the top level directory of the package but for ABINIT we prefer to do things in a much cleaner way using a build directory The advantage of this approach is that we keep object files and executables separated from the source code and this allows us to build different executables using the same source tree. For example, one can have a build directory with a version compiled with gfortran and another build directory for the intel ifort compiler or other builds done with same compiler but different compilation options.
Let’s call the build directory build_gfortran:
mkdir build_gfortran && cd build_gfortran
Now we should define the options that will be passed to the configure script. Instead of using the command line as done in the previous examples, we will be using an external file (myconf.ac9) to collect all our options. The syntax to read options from file is:
../configure --with-config-file="myconf.ac9"
where double quotation marks may be needed for portability reasons.
Note the use of ../configure as we are working inside the build directory build_gfortran while
the configure script is located in the top level directory of the package.
Important
The name of the options in myconf.ac9 is in normalized form that is
the initial -- is removed from the option name and all the other - characters in the string
are replaced by an underscore _.
Following these simple rules, the configure option --with-mpi becomes with_mpi in the ac9 file.
Also note that in the configuration file it is possible to use shell variables
and reuse the output of external tools using
backtick syntax
as is nf-config --flibs or, if you prefer, ${nf-config --flibs}.
This tricks allow us to reduce the amount of typing
and have configuration files that can be easily reused for other machines.
This is an example of configuration file in which we use the high-level interface
(with_LIBNAME=dirpath) as much as possible, except for linalg and FFTW3.
The explicit value of LIBNAME_LIBS and LIBNAME_FCFLAGS is also reported in the commented sections.
# -------------------------------------------------------------------------- #
# MPI support #
# -------------------------------------------------------------------------- #
# * the build system expects to find subdirectories named bin/, lib/,
# include/ inside the with_mpi directory
#
with_mpi=$HOME/local/
# Flavor of linear algebra libraries to use (default is netlib)
#
with_linalg_flavor="openblas"
# Library flags for linear algebra (default is unset)
#
LINALG_LIBS="-L$HOME/local/lib -lopenblas"
# -------------------------------------------------------------------------- #
# Optimized FFT support #
# -------------------------------------------------------------------------- #
# Flavor of FFT framework to support (default is auto)
#
# The high-level interface does not work yet so we pass options explicitly
#with_fftw3="$HOME/local/lib"
# Explicit options for fftw3
with_fft_flavor="fftw3"
FFTW3_LIBS="-L$HOME/local/lib -lfftw3f -lfftw3"
FFTW3_FCFLAGS="-L$HOME/local/include"
# -------------------------------------------------------------------------- #
# LibXC
# -------------------------------------------------------------------------- #
# Install prefix for LibXC (default is unset)
#
with_libxc="$HOME/local"
# Explicit options for libxc
#LIBXC_LIBS="-L$HOME/local/lib -lxcf90 -lxc"
#LIBXC_FCFLAGS="-I$HOME/local/include"
# -------------------------------------------------------------------------- #
# NetCDF
# -------------------------------------------------------------------------- #
# install prefix for NetCDF (default is unset)
#
with_netcdf=$(nc-config --prefix)
with_netcdf_fortran=$(nf-config --prefix)
# Explicit options for netcdf
#with_netcdf="yes"
#NETCDF_FORTRAN_LIBS=`nf-config --flibs`
#NETCDF_FORTRAN_FCFLAGS=`nf-config --fflags`
# install prefix for HDF5 (default is unset)
#
with_hdf5="$HOME/local"
# Explicit options for hdf5
#HDF5_LIBS=`nf-config --flibs`
#HDF5_FCFLAGS=`nf-config --fflags`
# Enable OpenMP (default is no)
enable_openmp="no"
A documented template with all the supported options can be found here
#
# Generic config file for ABINIT (documented template)
#
# After editing this file to suit your needs, you may save it as
# "~/.abinit/build/<hostname>.ac9" to keep these parameters as per-user
# defaults. Just replace "<hostname>" by the name of your machine,
# excluding the domain name.
#
# Example: if your machine is called "myhost.mydomain", you will save
# this file as "~/.abinit/build/myhost.ac9".
#
# You may put this file at the top level of an ABINIT source tree as well,
# in which case its definitions will apply to this particular tree only. In
# some situations, you may even want to put it at your current top build
# directory, in which case it will replace any other config file.
#
# Hint: If you do not know the name of your machine, just type "hostname"
# in a terminal, or "hostname -s" to obtain the name without domain name.
#
#
# IMPORTANT NOTES
#
# 1. Setting CPPFLAGS, CFLAGS, CXXFLAGS, or FCFLAGS manually is not
# recommended and will override any setting made by the build system.
# A gentler way to do is to use the CFLAGS_EXTRA, CXXFLAGS_EXTRA and
# FCFLAGS_EXTRA environment variables, or to override only one kind
# of flags. See the sections dedicated to C, C++ and Fortran below
# for details.
#
# 2. Do not forget to remove the leading "#" on a line when you customize
# an option.
#
# -------------------------------------------------------------------------- #
# Global build options #
# -------------------------------------------------------------------------- #
# Where to install ABINIT (default is /usr/local)
#
#prefix="${HOME}/hpc"
# Select debug level (default is basic)
#
# Allowed values:
#
# * none : strip debugging symbols
# * custom : allow for user-defined debug flags
# * basic : add '-g' option when the compiler allows for it
# * verbose : like basic + definition of the DEBUG_VERBOSE CPP option
# * enhanced : disable optimizations and debug verbosely
# * paranoid : enhanced debugging with additional warnings
# * naughty : paranoid debugging with array bound checks
#
# Levels other than no and yes are "profile mode" levels in which
# user-defined flags are overriden and optimizations disabled (see
# below)
#
# Note: debug levels are incremental, i.e. the flags of one level are
# appended to those of the previous ones
#
#with_debug_flavor="custom"
# Select optimization level whenever possible (default is standard,
# except when debugging is in profile mode - see above - in which case
# optimizations are turned off)
#
# Supported levels:
#
# * none : disable optimizations
# * custom : enable optimizations with user-defined flags
# * safe : build slow and very reliable code
# * standard : build fast and reliable code
# * aggressive : build very fast code, regardless of reliability
#
# Levels other than no and yes are "profile mode" levels in which
# user-defined flags are overriden
#
# Note:
#
# * this option is ignored when the debug is level is higher than basic
#
#with_optim_flavor="aggressive"
# Reduce AVX optimizations in sensitive subprograms (default is no)
#
#enable_avx_safe_mode="yes"
# Enable compiler hints (default is yes)
#
# Allowed values:
#
# * no : do not apply any hint
# * yes : apply all available hints
#
#enable_hints="no"
# -------------------------------------------------------------------------- #
# C support #
# -------------------------------------------------------------------------- #
# C preprocessor (should not be set in most cases)
#
#CPP="/usr/bin/cpp"
# C preprocessor custom debug flags (when with_debug_flavor=custom)
#
#CPPFLAGS_DEBUG="-DDEV_MG_DEBUG_MODE"
# C preprocessor custom optimization flags (when with_optim_flavor=custom)
#
#CPPFLAGS_OPTIM="-DDEV_DIAGO_DP"
# C preprocessor additional custom flags
#
#CPPFLAGS_EXTRA="-P"
# Forced C preprocessor flags
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#CPPFLAGS="-P"
# ------------------------------ #
# C compiler
#
#CC="gcc"
# C compiler custom debug flags (when with_debug_flavor=custom)
#
#CFLAGS_DEBUG="-g3"
# C compiler custom optimization flags (when with_optim_flavor=custom)
#
#CFLAGS_OPTIM="-O5"
# C compiler additional custom flags
#
#CFLAGS_EXTRA="-O2"
# Forced C compiler flags
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#CFLAGS="-O2"
# ------------------------------ #
# C linker custom debug flags (when with_debug_flavor=custom)
#
#CC_LDFLAGS_DEBUG="-Wl,-debug"
# C linker custom optimization flags (when with_optim_flavor=custom)
#
#CC_LDFLAGS_OPTIM="-Wl,-ipo"
# C linker additional custom flags
#
#CC_LDFLAGS_EXTRA="-Bstatic"
# Forced C linker flags
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#CC_LDFLAGS="-Bstatic"
# C linker custom debug libraries (when with_debug_flavor=custom)
#
#CC_LIBS_DEBUG="-ldebug"
# C linker custom optimization libraries (when with_optim_flavor=custom)
#
#CC_LIBS_OPTIM="-lopt_funcs"
# C linker additional custom libraries
#
#CC_LIBS_EXTRA="-lrt"
# Forced C linker libraries
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#CC_LIBS="-lrt"
# -------------------------------------------------------------------------- #
# C++ support #
# -------------------------------------------------------------------------- #
# Note: the XPP* environment variables will likely have no effect
# C++ preprocessor (should not be set in most cases)
#
#XPP="/usr/bin/cpp"
# C++ preprocessor custom debug flags (when with_debug_flavor=custom)
#
#XPPFLAGS_DEBUG="-DDEV_MG_DEBUG_MODE"
# C++ preprocessor custom optimization flags (when with_optim_flavor=custom)
#
#XPPFLAGS_OPTIM="-DDEV_DIAGO_DP"
# C++ preprocessor additional custom flags
#
#XPPFLAGS_EXTRA="-P"
# Forced C++ preprocessor flags
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#XPPFLAGS="-P"
# ------------------------------ #
# C++ compiler
#
#CXX="g++"
# C++ compiler custom debug flags (when with_debug_flavor=custom)
#
#CXXFLAGS_DEBUG="-g3"
# C++ compiler custom optimization flags (when with_optim_flavor=custom)
#
#CXXFLAGS_OPTIM="-O5"
# C++ compiler additional custom flags
#
#CXXFLAGS_EXTRA="-O2"
# Forced C++ compiler flags
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#CXXFLAGS="-O2"
# ------------------------------ #
# C++ linker custom debug flags (when with_debug_flavor=custom)
#
#CXX_LDFLAGS_DEBUG="-Wl,-debug"
# C++ linker custom optimization flags (when with_optim_flavor=custom)
#
#CXX_LDFLAGS_OPTIM="-Wl,-ipo"
# C++ linker additional custom flags
#
#CXX_LDFLAGS_EXTRA="-Bstatic"
# Forced C++ linker flags
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#CXX_LDFLAGS="-Bstatic"
# C++ linker custom debug libraries (when with_debug_flavor=custom)
#
#CXX_LIBS_DEBUG="-ldebug"
# C++ linker custom optimization libraries (when with_optim_flavor=custom)
#
#CXX_LIBS_OPTIM="-lopt_funcs"
# C++ linker additional custom libraries
#
#CXX_LIBS_EXTRA="-lblitz"
# Forced C++ linker libraries
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#CXX_LIBS="-lblitz"
# -------------------------------------------------------------------------- #
# Fortran support #
# -------------------------------------------------------------------------- #
# Fortran preprocessor (should not be set in most cases)
#
#FPP="/usr/local/bin/fpp"
# Fortran preprocessor custom debug flags (when with_debug_flavor=custom)
#
#FPPFLAGS_DEBUG="-DDEV_MG_DEBUG_MODE"
# Fortran preprocessor custom optimization flags (when with_optim_flavor=custom)
#
#FPPFLAGS_OPTIM="-DDEV_DIAGO_DP"
# Fortran preprocessor additional custom flags
#
#FPPFLAGS_EXTRA="-P"
# Forced Fortran preprocessor flags
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#FPPFLAGS="-P"
# ------------------------------ #
# Fortran compiler
#
#FC="gfortran"
# Fortran 77 compiler (addition for the Windows/Cygwin environment)
#
#F77="gfortran"
# Fortran compiler custom debug flags (when with_debug_flavor=custom)
#
#FCFLAGS_DEBUG="-g3"
# Fortran compiler custom OpenMP flags
#
#FCFLAGS_OPENMP="-fopenmp"
# Fortran compiler custom OpenMP GPU offload flags
#
#FCFLAGS_OPENMP_OFFLOAD="-fopenmp"
# Fortran compiler custom optimization flags (when with_optim_flavor=custom)
#
#FCFLAGS_OPTIM="-O5"
# Fortran compiler additional custom flags
#
#FCFLAGS_EXTRA="-O2"
# Forced Fortran compiler flags
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#FCFLAGS="-O2"
# Fortran flags for fixed-form source files
#
#FCFLAGS_FIXEDFORM="-ffixed-form"
# Fortran flags for free-form source files
#
#FCFLAGS_FREEFORM="-ffree-form"
# Fortran compiler flags to use a module directory
#
#FCFLAGS_MODDIR=="-J$(abinit_moddir)"
# Tricky Fortran compiler flags
#
#FCFLAGS_HINTS="-ffree-line-length-none"
# Fortran linker custom debug flags (when with_debug_flavor=custom)
#
#FC_LDFLAGS_DEBUG="-Wl,-debug"
# Fortran linker custom optimization flags (when with_optim_flavor=custom)
#
#FC_LDFLAGS_OPTIM="-Wl,-ipo"
# Fortran linker custom flags
#
#FC_LDFLAGS_EXTRA="-Bstatic"
# Forced Fortran linker flags
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#FC_LDFLAGS="-Bstatic"
# Fortran linker custom debug libraries (when with_debug_flavor=custom)
#
#FC_LIBS_DEBUG="-ldebug"
# Fortran linker custom optimization libraries (when with_optim_flavor=custom)
#
#FC_LIBS_OPTIM="-lopt_funcs"
# Fortran linker additional custom libraries
#
#FC_LIBS_EXTRA="-lsvml"
# Forced Fortran linker libraries
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#FC_LIBS="-lsvml"
# ------------------------------ #
# Use C clock instead of Fortran clock for timings (default is no)
#
#enable_cclock="yes"
# Wrap Fortran compiler calls (default is auto-detected)
# Combine this option with with_debug_flavor="basic" (or some other debug flavor) to keep preprocessed source
# files (they are removed by default, except if their build fails)
#
#enable_fc_wrapper="yes"
# Choose whether to read file lists from standard input or "ab.files"
# (default is yes = standard input)
#
#enable_stdin="no"
# Set per-directory Fortran optimizations (useful when a Fortran compiler
# crashes during the build)
#
# Note: this option is not available through the command line
#
#fcflags_opt_95_drive="-O0"
# -------------------------------------------------------------------------- #
# Python support #
# -------------------------------------------------------------------------- #
# Flags to pass to the Python interpreter (default is unset)
#
#PYFLAGS="-B"
# Preprocessing flags for C/Python bindings
#
#PYTHON_CPPFLAGS="-I/usr/local/include/numpy"
# -------------------------------------------------------------------------- #
# Libraries and linking #
# -------------------------------------------------------------------------- #
# Set archiver name
#
#AR="xiar"
# Archiver custom debug flags (when with_debug_flavor=custom)
#
#ARFLAGS_DEBUG=""
# Archiver custom optimization flags (when with_optim_flavor=custom)
#
#ARFLAGS_OPTIM=""
# Archiver additional custom flags
#
#ARFLAGS_EXTRA="-X 64"
# Forced archiver flags
# Note: will override build-system configuration - USE AT YOUR OWN RISKS!
#
#ARFLAGS="-X 32_64"
# ------------------------------ #
# Note: the following definitions are necessary for MINGW/WINDOW$ only
# and should be left unset on other architectures
# Archive index generator
#RANLIB="ranlib"
# Object symbols lister
#NM="nm"
# Generic linker
#LD="ld"
# Language-independent libraries to add to the build configuration
#LIBS="-lstdc++ -lpython2.7"
# -------------------------------------------------------------------------- #
# MPI support #
# -------------------------------------------------------------------------- #
# Determine whether to build parallel code (default is auto)
#
# Permitted values:
#
# * no : disable MPI support
# * yes : enable MPI support, assuming the compiler is MPI-aware
# * <prefix> : look for MPI in the <prefix> directory
#
# If left unset, the build system will take all appropriate decisions by
# itself, and MPI will be enabled only if the build environment supports
# it. If set to "yes", the configure script will stop if it does not find
# a working MPI environment.
#
# Note:
#
# * the build system expects to find subdirectories named bin/, lib/,
# include/ under the prefix.
#
#with_mpi="/usr/local/openmpi-gcc"
# Define MPI flavor (optional, only useful on some systems)
#
# Permitted values:
#
# * auto : Let the build system configure MPI (recommended).
#
# * double-wrap : Internally wrap the MPI compiler wrappers, only for
# severely bugged and oldish MPI implementations.
#
# * flags : Do not look for MPI compiler wrappers, only use
# compiler flags to enable MPI.
#
# * native : Assume that the compilers have native MPI support.
#
# * prefix : Assume that MPI wrappers are located under
# specified prefix and exclude any other
# configuration.
#
#with_mpi_flavor="native"
# Activate code branches to perform MPI operations on GPU buffers,
# which allow performance gains on intra-node communications.
# WARNING: this feature requires a MPI that is "GPU-aware", which usually
# means "compiled with CUDA/ROCm support"
#
#enable_mpi_gpu_aware="no"
# Activate the MPI_IN_PLACE option whenever possible (default is no)
# WARNING: this feature requires MPI2, ignored if the MPI library
# is not MPI2 compliant.
#
#enable_mpi_inplace="no"
# Activate parallel I/O (default is auto)
#
# Permitted values:
#
# * auto : let the configure script auto-detect MPI I/O support
# * no : disable MPI I/O support
# * yes : enable MPI I/O support
#
# If left unset, the build system will take all appropriate decisions by
# itself, and MPI I/O will be enabled only if the build environment supports
# it. If set to "yes", the configure script will stop if it does not find
# a working MPI I/O environment.
#
#enable_mpi_io="yes"
# Enable MPI-IO mode in Abinit (use MPI I/O as default I/O library,
# change the default values of iomode)
# Beware that not all the features of Abinit support MPI I/O,
# This options is mainly used by developers for debugging purposes.
#
#enable_mpi_io_default="no"
# Set MPI standard level (default is auto-detected)
# Note: all current implementations should support at least level 2
#
# Supported levels:
#
# * 1 : use 'mpif.h' header
# * 2 : use mpi Fortran module
# * 3 : use mpi_f08 Fortran module
#
#with_mpi_level="2"
# C preprocessor flags for MPI (default is unset)
#
#MPI_CPPFLAGS="-I/usr/local/include"
# C flags for MPI (default is unset)
#
#MPI_CFLAGS=""
# C++ flags for MPI (default is unset)
#
#MPI_CXXFLAGS=""
# Fortran flags for MPI (default is unset)
#
#MPI_FCFLAGS=""
# Link flags for MPI (default is unset)
#
#MPI_LDFLAGS=""
# Library flags for MPI (default is unset)
#
#MPI_LIBS="-L/usr/local/lib -lmpi"
# -------------------------------------------------------------------------- #
# GPU support #
# -------------------------------------------------------------------------- #
# Requirement: go through README.GPU before doing anything
#
# Note: this is highly experimental - USE AT YOUR OWN RISKS!
# Trigger and install prefix for NVIDIA CUDA libraries and compilers
#
# Permitted values:
#
# * no : disable CUDA support
# * yes : enable CUDA support, assuming the build environment
# is properly set
# * <prefix> : look for CUDA in the <prefix> directory
#
# Note: The build system expects to find subdirectories named bin/, lib/,
# include/ under the prefix.
#
#with_cuda="/usr/local/cuda"
# CUDA C preprocessor flags (default is unset)
#
#CUDA_CPPFLAGS="-I/usr/local/include/cuda"
# CUDA C flags (default is unset)
#
#CUDA_CFLAGS="-I/usr/local/include/cuda"
# CUDA C++ flags (default is unset)
#
#CUDA_CXXFLAGS="-std=c++11"
# CUDA Fortran flags (default is unset)
#
#CUDA_FCFLAGS="-I/usr/local/include/cuda"
# CUDA link flags (default is unset)
#
#CUDA_LDFLAGS="-L/usr/local/cuda/lib64 -lcublas -lcufft -lcudart"
# CUDA link flags (default is unset)
#
#CUDA_LIBS="-L/usr/local/cuda/lib64 -lcublas -lcufft -lcudart"
# Trigger and install prefix for AMD ROCM libraries and compilers
#
# Permitted values:
#
# * no : disable ROCM support
# * yes : enable ROCM support, assuming the build environment
# is properly set
# * <prefix> : look for ROCM in the <prefix> directory
#
# Note: The build system expects to find subdirectories named bin/, lib/,
# include/ under the prefix.
#
#with_rocm="/opt/rocm"
# ROCM C preprocessor flags (default is unset)
#
#ROCM_CPPFLAGS="-I/usr/local/include/cuda"
# ROCM C flags (default is unset)
#
#ROCM_CFLAGS="-I/usr/local/include/cuda"
# ROCM C++ flags (default is unset)
#
#ROCM_CXXFLAGS="-std=c++11"
# ROCM Fortran flags (default is unset)
#
#ROCM_FCFLAGS="-I/usr/local/include/cuda"
# ROCM link flags (default is unset)
#
#ROCM_LDFLAGS="-L/usr/local/cuda/lib64 -lcublas -lcufft -lcudart"
# ROCM link flags (default is unset)
#
#ROCM_LIBS="-L/usr/local/cuda/lib64 -lcublas -lcufft -lcudart"
# Trigger and install prefix for GPU (CUDA or ROCM) libraries and compilers - deprecated
#
# Permitted values:
#
# * no : disable GPU support
# * yes : enable GPU support, assuming the build environment
# is properly set
# * <prefix> : look for GPU in the <prefix> directory
#
# Note: The build system expects to find subdirectories named bin/, lib/,
# include/ under the prefix.
#
#with_gpu="/usr/local/cuda"
# Flavor of the GPU library to use (default is cuda-single) - deprecated
#
# Supported libraries:
#
# * cuda-single : Cuda with single-precision arithmetic
# * cuda-double : Cuda with double-precision arithmetic
# * hip-double : HIP with double-precision arithmetic
# * none : not implemented (will replace enable_gpu)
#
#with_gpu_flavor="cuda-double"
# GPU C preprocessor flags (default is unset) - deprecated
#
#GPU_CPPFLAGS="-I/usr/local/include/cuda"
# GPU C flags (default is unset) - deprecated
#
#GPU_CFLAGS="-I/usr/local/include/cuda"
# GPU C++ flags (default is unset) - deprecated
#
#GPU_CXXFLAGS="-std=c++11"
# GPU Fortran flags (default is unset) - deprecated
#
#GPU_FCFLAGS="-I/usr/local/include/cuda"
# GPU link flags (default is unset) - deprecated
#
#GPU_LDFLAGS="-L/usr/local/cuda/lib64 -lcublas -lcufft -lcudart"
# GPU link flags (default is unset) - deprecated
#
#GPU_LIBS="-L/usr/local/cuda/lib64 -lcublas -lcufft -lcudart"
# GPU arch, useful for autosetting OpenMP offload flags (default is NVIDIA Ampere)
# Example values : - 60 (NVIDIA Pascal)
# - 70 (NVIDIA Volta)
# - 75 (NVIDIA Turing)
# - 80 (NVIDIA Ampere)
# - 90 (NVIDIA Hopper)
# - gfx90a (AMD Instinct MI250)
#
#GPU_ARCH=80
# Enable Unified Memory for NVIDIA GPUs (exclusive to NVHPC)
#
#enable_gpu_nvidia_unified_memory="no"
# ------------------------------ #
# Advanced GPU options (experts only)
#
# DO NOT EDIT THIS SECTION UNLESS YOU *TRULY* KNOW WHAT YOU ARE DOING!
# In any case, the outcome of setting the following options is highly
# impredictible.
# nVidia C compiler (should not be set)
#
#NVCC="/usr/local/cuda/bin/nvcc"
# Forced nVidia C compiler preprocessing flags (should not be set)
#
#NVCC_CPPFLAGS="-DHAVE_CUDA_SDK"
# Forced nVidia C compiler flags (should not be set)
#
#NVCC_CFLAGS="-O3 -Xptxas=-v --use_fast_math --compiler-options -O3,-fPIC -DHAVE_CONFIG_H --forward-unknown-opts"
# Forced nVidia C compiler flags specific to arch (should not be set)
#
#NVCC_CFLAGS_ARCH="-arch=sm_13"
# Forced nVidia linker flags (should not be set)
#
#NVCC_LDFLAGS=""
# Forced nVidia linker libraries (should not be set)
#
#NVCC_LIBS=""
# -------------------------------------------------------------------------- #
# Linear algebra support #
# -------------------------------------------------------------------------- #
# A large set of linear algebra libraries are available on the Web.
# See e.g. https://hpc.llnl.gov/manuals/mathematical-software/math-libraries-and-interactive-tools
# Among all those, many are supported by ABINIT.
# WARNING: when setting the value of the linear algebra flavor to "custom",
# the associated CPP options may be defined in an impredictable
# manner by the build system. In such a case, checking which
# CPP options are enabled by looking at the output of abinit
# is thus highly recommended before running production
# calculations.
# Supported libraries:
#
# * aocl : AMD Optimizing CPU Libraries
# https://developer.amd.com/amd-aocl/
#
# * atlas : Automatically Tuned Linear Algebra Software
# http://math-atlas.sourceforge.net/
#
# * auto : automatically look for linear algebra libraries
# depending on the build environment (default)
#
# * easybuild : EasyBuild - Building software with ease
# https://easybuilders.github.io/easybuild/
#
# * elpa : Eigen soLvers for Petaflops Applications
# http://elpa.rzg.mpg.de
#
# * magma : Matrix Algebra on GPU and Multicore Architectures
# (MAGMA minimal version>=1.1.0, requires Cuda)
# http://icl.cs.utk.edu/magma/
#
# * mkl : Intel Math Kernel Library
# http://software.intel.com/en-us/articles/intel-mkl/
#
# * netlib : Netlib repository reference libraries
# http://www.netlib.org/lapack/
# http://www.netlib.org/scalapack/
#
# * none : just check user-specified libraries, do not try to
# detect their type
#
#
# * nvpl : Nvidia Performance Library (NVPL)
# (only for ARM processors)
# https://docs.nvidia.com/nvpl/
#
# * openblas : OpenBLAS - An Optimized BLAS Library
# https://www.openblas.net/
#
# * plasma : Parallel Linear Algebra for Scalable Multicore
# Architectures (requires MPI)
# http://icl.cs.utk.edu/plasma/
#
# Notes:
#
# * you may combine "magma" and/or "plasma" with
# any other flavor, using '+' as a separator
#
# * "custom" also works when the Fortran compiler provides a full
# BLAS+LAPACK implementation internally (e.g. Lahey Fortran)
#
# * the include and link flags for MAGMA and ScaLAPACK have to be
# specified together with those of BLAS and LAPACK (see options below)
#
# * please consult the MKL link line advisor if you experience
# problems with MKL, by going to
# http://software.intel.com/en-us/articles/intel-mkl-link-line-advisor/
#
# * if with_linalg is set to "no", linear algebra tests will be disabled
# and the configuration will be assumed to work as-is by the build
# system (USE AT YOUR OWN RISKS)
#
# * with_linalg_incs and with_linalg_libs systematically override the
# contents of with_linalg
#
# * in order to enable SCALAPACK, you might have to specify -lscalapack
# or use another appropriate syntax for your system.
#
# Install prefix for linear algebra
#
#with_linalg="/usr/local"
# Flavor of linear algebra libraries to use (default is netlib)
#
#with_linalg_flavor="openblas"
# C preprocessing flags for linear algebra (default is unset)
#
#LINALG_CPPFLAGS="-I/usr/local/include"
# C flags for linear algebra (default is unset)
#
#LINALG_CFLAGS="-m64"
# C++ flags for linear algebra (default is unset)
#
#LINALG_CXXFLAGS="-m64"
# Fortran flags for linear algebra (default is unset)
#
#LINALG_FCFLAGS="-I/usr/local/include"
# Link flags for linear algebra (default is unset)
#
#LINALG_LDFLAGS=""
# Library flags for linear algebra (default is unset)
#
#LINALG_LIBS="-L/usr/local/lib -llapack -lblas -lscalapack"
# -------------------------------------------------------------------------- #
# Optimized FFT support #
# -------------------------------------------------------------------------- #
# Supported libraries:
#
# * auto : select library depending on build environment (default)
# * custom : bypass build-system checks
# * dfti : native MKL FFT library
# * fftw3 : serial FFTW3 library
# * fftw3-threads : threaded FFTW3 library
# * nvpl : NVPL: Nvidia Performance Library (only for ARM)
# * pfft : MPI-parallel PFFT library (for maintainers only)
# * goedecker : Abinit internal FFT
#
# Notes:
#
# * only one flavor can be selected at a time, flavors being mutually
# exclusive
#
# The following lines relate to a generic FFT library (e.g. they are used for dfti)
# Install prefix for the FFT library
#
#with_fft="/usr/local"
# Flavor of FFT framework to support (default is auto)
#
#with_fft_flavor="fftw3"
# C preprocessor flags for the FFT framework (default is unset)
#
#FFT_CPPFLAGS="-I/usr/local/include/fftw"
# C flags for the FFT framework (default is unset)
#
#FFT_CFLAGS="-I/usr/local/include/fftw"
# Fortran flags for the FFT framework (default is unset)
#
#FFT_FCFLAGS="-I/usr/local/include/fftw"
# Link flags for the FFT framework (default is unset)
#
#FFT_LDFLAGS="-L/usr/local/lib/fftw -lfftw3"
# Library flags for the FFT framework (default is unset)
#
#FFT_LIBS="-L/usr/local/lib/fftw -lfftw3"
# ------------------------------ #
# The following lines relate specifically to the FFTW3 library
# Install prefix for the FFTW3 library
#
#with_fftw3="/usr/local"
# C preprocessor flags for the FFTW3 library (default is unset)
#
#FFTW3_CPPFLAGS="-I/usr/local/include/fftw"
# C flags for the FFTW3 library (default is unset)
#
#FFTW3_CFLAGS="-I/usr/local/include/fftw"
# Fortran flags for the FFTW3 library (default is unset)
#
#FFTW3_FCFLAGS="-I/usr/local/include/fftw"
# Link flags for the FFTW3 library (default is unset)
#
#FFTW3_LDFLAGS="-L/usr/local/lib/fftw -lfftw3"
# Library flags for the FFTW3 library (default is unset)
#
#FFTW3_LIBS="-L/usr/local/lib/fftw -lfftw3"
# ------------------------------ #
# The following lines relate specifically to the PFFT library
# Install prefix for the PFFT library
#
#with_pfft="/usr/local"
# C preprocessor flags for the PFFT library (default is unset)
#
#PFFT_CPPFLAGS="-I/usr/local/include/fftw"
# C flags for the PFFT library (default is unset)
#
#PFFT_CFLAGS="-I/usr/local/include/fftw"
# Fortran flags for the PFFT library (default is unset)
#
#PFFT_FCFLAGS="-I/usr/local/include/fftw"
# Link flags for the PFFT library (default is unset)
#
#PFFT_LDFLAGS="-L/usr/local/lib/fftw -lfftw3"
# Library flags for the PFFT library (default is unset)
#
#PFFT_LIBS="-L/usr/local/lib/fftw -lfftw3"
# -------------------------------------------------------------------------- #
# Feature triggers #
# -------------------------------------------------------------------------- #
# Through feature triggers, the build system of Abinit tries to link
# prioritarily with external libraries to provide the requested
# functionality. When unsuccessful, Abinit will run in degraded mode,
# which means that it will provide poor performance and scalability, as
# well as refuse to run some standard calculations. However, in some
# cases, for historical reasons, it can resort to a limited internal
# implementation.
#
# Enabling feature triggers is necessary for packaging and is recommended
# in most other cases. Relying upon external optimized libraries is always
# smarter than embedding their source code, as their performance and
# integration within the local environment are always significantly
# better.
#
# The following optional dependencies are sorted by alphabetical order.
# Please note that some of them may depend on others, as indicated.
# Notes:
#
# * when specifying with_package="prefix", the build system automatically
# looks for the relevant libraries in prefix/include and prefix/lib
#
# * the 'with_package' options can also be set to yes or no, to let the
# build system find out the corresponding parameters on plaftorms where
# the external packages are available system-wide
#
# * the 'with_package_incs' and 'with_package_libs' options systematically
# override the settings provided by corresponding 'with_package' options
#
# ------------------------------ #
# BigDFT (depends on LibXC, see below)
# Website: http://bigdft.org/
# Enable BigDFT support (default is no)
#
#with_bigdft="yes"
# Fortran flags for BigDFT (default is unset)
#
#BIGDFT_FCFLAGS="-I/usr/local/include/bigdft"
# Link flags for BigDFT (default is unset)
#
#BIGDFT_LDFLAGS=""
# Library flags for BigDFT (default is unset)
#
#BIGDFT_LIBS="-L/usr/local/lib/bigdft -lbigdft -lpoissonsolver -labinit"
# ------------------------------ #
# Kokkos Core library
# Website: http://kokkos.org/
# Enable Kokkos Core support (default is no)
#
#with_kokkos="yes"
# C++ flags for Kokkos Core (default is unset)
#
#KOKKOS_CXXFLAGS="-I/usr/local/include/kokkos"
# Fortran flags for Kokkos Core (default is unset)
#
#KOKKOS_FCFLAGS="-I/usr/local/include/kokkos"
# Link flags for Kokkos Core (default is unset)
#
#KOKKOS_LDFLAGS=""
# Library flags for Kokkos Core (default is unset)
#
#KOKKOS_LIBS="-L/usr/local/lib/kokkos -lkokkoscore"
# ------------------------------ #
# Levenberg-Marquardt algorithm
# Website: http://users.ics.forth.gr/~lourakis/levmar/
# Trigger and install prefix for Levmar (default is unset)
#
#with_levmar="yes"
# C preprocessor flags for Levmar (default is unset)
#
#LEVMAR_CPPFLAGS="-I/opt/etsf/include"
# C flags for Levmar (default is unset)
#
#LEVMAR_CFLAGS="-I/opt/etsf/include"
# Fortran flags for Levmar (default is unset)
#
#LEVMAR_FCFLAGS="-I/opt/etsf/include"
# Link flags for Levmar (default is unset)
#
#LEVMAR_LDFLAGS=""
# Library flags for Levmar (default is unset)
#
#LEVMAR_LIBS="-L/opt/etsf/lib -llevmar"
# ------------------------------ #
# LibXC
# Website: http://www.tddft.org/programs/octopus/wiki/index.php/Libxc
# Trigger and install prefix for LibXC (default is unset)
#
#with_libxc="/usr/local"
# C preprocessor flags for LibXC (default is unset)
#
#LIBXC_CPPFLAGS="-I/usr/local/include"
# C flags for LibXC (default is unset)
#
#LIBXC_CFLAGS="-I/usr/local/include"
# Fortran flags for LibXC (default is unset)
#
#LIBXC_FCFLAGS="-I/usr/local/include"
# Link flags for the LibXC library (default is unset)
#
#LIBXC_LDFLAGS=""
# Library flags for LibXC (default is unset)
#
#LIBXC_LIBS="-L/usr/local/lib -lxcf90 -lxc"
# ------------------------------ #
# LibXML2
# Website: http://www.xmlsoft.org/
# Trigger and install prefix for LibXML2 (default is unset)
#
#with_libxml2="/usr/local"
# C preprocessor flags for LibXML2 (default is unset)
#
#LIBXML2_CPPFLAGS="-I/usr/local/include"
# C flags for LibXML2 (default is unset)
#
#LIBXML2_CFLAGS="-I/usr/local/include"
# Fortran flags for LibXML2 (default is unset)
#
#LIBXML2_FCFLAGS="-I/usr/local/include"
# Link flags for the LibXML2 library (default is unset)
#
#LIBXML2_LDFLAGS=""
# Library flags for LibXML2 (default is unset)
#
#LIBXML2_LIBS="-L/usr/local/lib -lxcf90 -lxc"
# ------------------------------ #
# HDF5
# Website: https://www.hdfgroup.org/
# Trigger and install prefix for HDF5 (default is unset)
#
#with_hdf5="/usr/local"
# Location of the h5cc (or h5pcc) compiler (default is unset)
#
# Note: This parameter can greatly help the build system of ABINIT in
# selecting the correct version of HDF5.
#
#H5CC="/opt/local/hdf5-serial/bin/h5cc"
# C preprocessing flags for HDF5 (default is unset)
#
#HDF5_CPPFLAGS="-I/usr/local/include/hdf5"
# C flags for HDF5 (default is unset)
#
#HDF5_CFLAGS="-std=c99"
# Fortran flags for HDF5 (default is unset)
#
#HDF5_FCFLAGS="-I/usr/local/include/hdf5"
# Link flags for HDF5 (default is unset)
#
#HDF5_LDFLAGS=""
# Library flags for HDF5 (default is unset)
#
#HDF5_LIBS="-L/usr/local/lib/hdf5 -lhdf5 -lhdf5_hl"
# ------------------------------ #
# NetCDF
# Website: http://www.unidata.ucar.edu/software/netcdf/
# Trigger and install prefix for NetCDF (default is unset)
#
#with_netcdf="/usr/local"
# C preprocessing flags for NetCDF (default is unset)
#
#NETCDF_CPPFLAGS="-I/usr/local/include/netcdf"
# C flags for NetCDF (default is unset)
#
#NETCDF_CFLAGS="-std=c99"
# Fortran flags for NetCDF (default is unset)
#
#NETCDF_FCFLAGS="-I/usr/local/include/netcdf"
# Link flags for NetCDF (default is unset)
#
#NETCDF_LDFLAGS=""
# Library flags for NetCDF (default is unset)
#
#NETCDF_LIBS="-L/usr/local/lib/netcdf -lnetcdf"
# Enable Netcdf mode in Abinit (use netcdf as default I/O library,
# change the default values of accesswff)
#
#enable_netcdf_default="no"
# ------------------------------ #
# NetCDF-Fortran
# Website: http://www.unidata.ucar.edu/software/netcdf/
# Trigger and install prefix for NetCDF-Fortran (default is unset)
#
#with_netcdf_fortran="/usr/local"
# C preprocessing flags for NetCDF-Fortran (default is unset)
#
#NETCDF_FORTRAN_CPPFLAGS="-I/usr/local/include/netcdf_fortran"
# C flags for NetCDF-Fortran (default is unset)
#
#NETCDF_FORTRAN_CFLAGS=""
# Fortran flags for NetCDF-Fortran (default is unset)
#
#NETCDF_FORTRAN_FCFLAGS="-I/usr/local/include/netcdf_fortran"
# Link flags for NetCDF-Fortran (default is unset)
#
#NETCDF_FORTRAN_LDFLAGS=""
# Library flags for NetCDF-Fortran (default is unset)
#
#NETCDF_FORTRAN_LIBS="-L/usr/local/lib/netcdf_fortran -lnetcdff"
# ------------------------------ #
# Enable LibPSML support (default is no)
#
#with_libpsml="yes"
# Fortran flags for the LibPSML library (default is unset)
#
#LIBPSML_FCFLAGS="-I/usr/local/libpsml-1.1.7/include"
# Link flags for the LibPSML library (default is unset)
#
#LIBPSML_LDFLAGS=""
# Library flags for LibPSML (default is unset)
#
#LIBPSML_LIBS="-L/usr/local/libpsml-1.1.7/lib -lpsml"
# ------------------------------ #
# TRIQS DMFT library (for CTQMC and rotationally invariant interaction)
# Website: https://triqs.ipht.cnrs.fr/1.x/index.html
# Trigger and install prefix for TRIQS (default is unset)
#
#with_triqs="/usr/local"
# Allow Fortran programs to call Python
#
#enable_python_invocation="yes"
# Select the 1.4 version of TRIQS
#
#enable_triqs_v1_4="yes"
# Select the 2.0 version of TRIQS
#
#enable_triqs_v2_0="yes"
# Select the complex version of TRIQS
#
#enable_triqs_complex="yes"
# C++ flags for the TRIQS library (default is unset)
#
#TRIQS_CXXFLAGS="-I/usr/local/triqs/include -I/usr/local/cthyb/src/c++"
# Fortran flags for the TRIQS library (default is unset)
#
#TRIQS_FCFLAGS="-I/usr/local/triqs/include -I/usr/local/cthyb/src/c++"
# Link flags for the TRIQS library (default is unset)
#
#TRIQS_LDFLAGS="-L/usr/local/triqs/lib -ltriqs -lcthyb_c"
# Link flags for the TRIQS library (default is unset)
#
#TRIQS_LIBS="-L/usr/local/triqs/lib -ltriqs -lcthyb_c"
# ------------------------------ #
# Enable Wannier90 support (default is no)
#
#with_wannier90="yes"
# Fortran flags for the Wannier90 library (default is unset)
#
#WANNIER90_FCFLAGS="-I/usr/local/include/wannier90"
# Link flags for the Wannier90 library (default is unset)
#
#WANNIER90_LDFLAGS=""
# Link flags for the Wannier90 library (default is unset)
#
#WANNIER90_LIBS="-L${HOME}/lib/wannier90 -lwannier90"
# ------------------------------ #
# Enable XMLF90 support (default is no)
#
#with_xmlf90="yes"
# Fortran flags for the XMLF90 library (default is unset)
#
#XMLF90_FCFLAGS="-I/usr/local/include"
# Link flags for the LibPSML library (default is unset)
#
#XMLF90_LDFLAGS=""
# Link flags for the LibPSML library (default is unset)
#
#XMLF90_LIBS="-L/usr/local/lib -lxmlf90"
# ------------------------------ #
# YAKL (Yet Another Kernel Launcher) library
# Small C++ framework used in ABINIT for GPU memory pool allocation
# Website: https://github.com/mrnorman/YAKL
# Enable YAKL support (default is no)
#
#with_yakl="yes"
# Fortran flags for YAKL (default is unset)
#
#YAKL_FCFLAGS="-I/usr/local/include/yakl"
# Link flags for YAKL (default is unset)
#
#YAKL_LDFLAGS=""
# Library flags for YAKL (default is unset)
#
#YAKL_LIBS="-L/usr/local/lib/yakl -lyakl_fortran_interface"
# -------------------------------------------------------------------------- #
# Profiling and performance analysis #
# -------------------------------------------------------------------------- #
# Enable internal Abinit timer (default is yes)
#
#enable_timer="no"
# Enable PAPI support (default is no)
#
#with_papi="yes"
# C preprocessor flags for the PAPI library (default is unset)
#
#PAPI_CPPFLAGS="-I/usr/local/include/papi"
# C flags for the PAPI library (default is unset)
#
#PAPI_CFLAGS="-I/usr/local/include/papi"
# Fortran flags for the PAPI library (default is unset)
#
#PAPI_FCFLAGS="-I/usr/local/include/papi"
# Link flags for the PAPI library (default is unset)
#
#PAPI_LDFLAGS=""
# Library flags for the PAPI library (default is unset)
#
#PAPI_LIBS="-L/usr/local/lib/papi -lpapi"
# -------------------------------------------------------------------------- #
# Fallbacks #
# -------------------------------------------------------------------------- #
# Fallbacks are external packages pre-built together for Abinit, in order to
# partly compensate for the absence of required external packages. If used,
# they will provide lower reliability, performance and scalability than the
# proper installation of the full-fledged corresponding packages. In any case,
# they should not be used in production.
#
# Fallbacks are enabled when the "with_fallbacks" option below points to
# an existing and readable directory. If this is not the case, failed
# detections of external packages will cause configure to abort.
#
# Example: if you set with_fallbacks="/path/to/fallbacks", the
# build system of Abinit will look for:
#
# * executables in /path/to/fallbacks/bin/
# * C headers and Fortran modules in /path/to/fallbacks/include/
# * libraries in /path/to/fallbacks/lib/
#
# when it cannot detect properly installed external packages.
#
# Please note that this mechanism will work only if you have built and
# installed the fallbacks you need beforehand. You can have a look at
# the following helper script after running configure for more
# information:
#
# ~abinit-builddir/fallbacks/build-abinit-fallbacks.sh
#
# Comprehensive documentation about how to use external packages properly
# and build fallbacks can be found on the Abinit Wiki, at:
#
# https://wiki.abinit.org/
#
# Prefix for already installed fallbacks (default is unset)
#
#with_fallbacks="/opt/abinit-dev/fallbacks/gnu/4.9/mpi"
# -------------------------------------------------------------------------- #
# Build workflow #
# -------------------------------------------------------------------------- #
# Note: if you do not know what the following is about, just leave it unset.
# Use external ABINIT Common (default is no)
#
#with_abinit_common="yes"
# Fortran flags for ABINIT Common (default is unset)
#
#ABINIT_COMMON_FCFLAGS="-I/usr/local/include/abinit_common"
# Link flags for ABINIT Common (default is unset)
#
#ABINIT_COMMON_LDFLAGS=""
# Library flags for ABINIT Common (default is unset)
#
#ABINIT_COMMON_LIBS="-L${HOME}/abinit/lib -labinit_common"
# ------------------------------ #
# Use external LibPAW (default is no)
#
#with_libpaw="yes"
# C preprocessing flags for LibPAW (default is unset)
#
#LIBPAW_CPPFLAGS="-I/usr/local/include/libpaw"
# C flags for LibPAW (default is unset)
#
#LIBPAW_CFLAGS="-std=c99"
# Fortran flags for LibPAW (default is unset)
#
#LIBPAW_FCFLAGS="-I/usr/local/include/libpaw"
# Link flags for LibPAW (default is unset)
#
#LIBPAW_LDFLAGS=""
# Library flags for LibPAW (default is unset)
#
#LIBPAW_LIBS="-L${HOME}/abinit_shared/lib -lpaw"
# -------------------------------------------------------------------------- #
# Developer options #
# -------------------------------------------------------------------------- #
# Note: all the following options are disabled by default because they
# enable the build of highly experimental code (i.e. they have
# the complementary default values to those displayed below)
#
# Uncomment the following lines at your own risks
# Enable optimized cRPA build for ifort <=17 (routerovitch)
#
#enable_crpa_optim="yes"
# Enable double precision for GW calculations (gmatteo)
#
#enable_gw_dpc="yes"
# Use tetrahedrons from Libtetra (torrent)
#
#enable_libtetra="yes"
# Enable lotf (mancini)
#
#enable_lotf="yes"
# Enable memory profiling (waroquiers)
#
#enable_memory_profiling="yes"
# Enable OpenMP (gmatteo, torrent)
#
#enable_openmp="yes"
# Enable OpenMP GPU offload (sarrautem, torrent)
#
#enable_openmp_offload="yes"
# Enable ZDOTC and ZDOTU bugfix (gmatteo)
#
#enable_zdot_bugfix="yes"
# Activate workaround for buggy MPI interfaces (not having interfaces for scalars)
#
#enable_mpi_interfaces_bugfix="yes"
# -------------------------------------------------------------------------- #
# Maintainer options #
# -------------------------------------------------------------------------- #
# Fortran compiler vendor to be used by the build system (default is unset)
#
# Note: do not use if you do not know what it is about
#
#with_fc_vendor="dummy"
# Fortran compiler version to be used by the build system (default is unset)
#
# Note: do not use if you do not know what it is about
#
#with_fc_version="0.0"
# Build debugging instructions present in the source code
#
#enable_source_debug="yes"
Copy the content of the example in myconf.ac9, then run:
../configure --with-config-file="myconf.ac9"
If everything goes smoothly, you should obtain the following summary:
==============================================================================
=== Final remarks ===
==============================================================================
Core build parameters
---------------------
* C compiler : gnu version 5.3
* Fortran compiler : gnu version 5.3
* architecture : intel xeon (64 bits)
* debugging : basic
* optimizations : standard
* OpenMP enabled : no (collapse: ignored)
* MPI enabled : yes (flavor: auto)
* MPI in-place : no
* MPI-IO enabled : yes
* GPU enabled : no (flavor: none)
* LibXML2 enabled : no
* LibPSML enabled : no
* XMLF90 enabled : no
* HDF5 enabled : yes (MPI support: yes)
* NetCDF enabled : yes (MPI support: yes)
* NetCDF-F enabled : yes (MPI support: yes)
* FFT flavor : fftw3 (libs: user-defined)
* LINALG flavor : openblas (libs: user-defined)
* Build workflow : monolith
0 deprecated options have been used:.
Configuration complete.
You may now type "make" to build Abinit.
(or "make -j<n>", where <n> is the number of available processors)
Important
Please take your time to read carefully the final summary and make sure you are getting what you expect. A lot of typos or configuration errors can be easily spotted at this level.
You might then find useful to have a look at other examples available in this page. Additional configuration files for clusters can be found in the abiconfig package.
The configure script has generated several Makefiles required by make as well as the config.h include file with all the pre-processing options that will be used to build ABINIT. This file is included in every ABINIT source file and it defines the features that will be activated or deactivated at compilation-time depending on the libraries available on your machine. Let’s have a look at a selected portion of config.h:
/* Define to 1 if you have a working MPI installation. */
#define HAVE_MPI 1
/* Define to 1 if you have a MPI-1 implementation (obsolete, broken). */
/* #undef HAVE_MPI1 */
/* Define to 1 if you have a MPI-2 implementation. */
#define HAVE_MPI2 1
/* Define to 1 if you want MPI I/O support. */
#define HAVE_MPI_IO 1
/* Define to 1 if you have a parallel NetCDF library. */
/* #undef HAVE_NETCDF_MPI */
This file tells us that
- we are building ABINIT with MPI support
- we have a library implementing the MPI2 specifications
- our MPI implementation supports parallel MPI-IO. Note that this does not mean that netcdf supports MPI-IO. In this example, indeed, HAVE_NETCDF_MPI is undefined and this means the library does not have parallel-IO capabilities.
Of course, end users are mainly concerned with the final summary reported
by the configure script to understand whether
a particular feature has been activated or not but more advanced users may
find the content of config.h valuable to understand what’s going on.
Now we can finally compile the package with e.g. make -j2. If the compilation completes successfully (🙌), you should end up with a bunch of executables inside src/98_main. Note, however, that the fact that the compilation completed successfully does not necessarily imply that the executables will work as expected as there are many different things that can go wrong at runtime.
First of all, let’s try to execute:
abinit --version
Tip
If this is a parallel build, you may need to use
mpirun -n 1 abinit --version
even for a sequential run as certain MPI libraries are not able to bootstrap the MPI library without mpirun (mpiexec). On some clusters with Slurm, the syadmin may ask you to use srun instead of mpirun.
To get the summary of options activated during the build, run abinit with the -b option
(or --build if you prefer the verbose version)
./src/98_main/abinit -b
If the executable does not crash (🙌), you may want to execute
make test_fast
to run some basic tests. If something goes wrong when executing the binary or when running the tests, checkout the Troubleshooting section for possible solutions.
Finally, you may want to execute the runtests.py python script in the tests directory in order to validate the build before running production calculations:
cd tests
../../tests/runtests.py v1 -j4
As usual, use:
../../tests/runtests.py --help
to list the available options. A more detailed discussion is given in this page.

Dynamic libraries and ldd¶
Since we decided to compile with dynamic linking, the external libraries are not included in the final executables.
Actually, the libraries will be loaded by the Operating System (OS) at runtime when we execute the binary.
The OS will search for dynamic libraries using the list of directories specified
in $LD_LIBRARY_PATH ($DYLD_LIBRARY_PATH for MacOs).
A typical mistake is to execute abinit with a wrong $LD_LIBRARY_PATH that is either empty or
different from the one used when compiling the code
(if it’s different and it works, I assume you know what you are doing so you should not be reading this section!)
On Linux, one can use the ldd tool to print the shared objects (shared libraries) required by each program or shared object specified on the command line:
ldd src/98_main/abinit
linux-vdso.so.1 (0x00007fffbe7a4000)
libopenblas.so.0 => /home/gmatteo/local/lib/libopenblas.so.0 (0x00007fc892155000)
libnetcdff.so.7 => /home/gmatteo/local/lib/libnetcdff.so.7 (0x00007fc891ede000)
libnetcdf.so.15 => /home/gmatteo/local/lib/libnetcdf.so.15 (0x00007fc891b62000)
libhdf5_hl.so.200 => /home/gmatteo/local/lib/libhdf5_hl.so.200 (0x00007fc89193c000)
libhdf5.so.200 => /home/gmatteo/local/lib/libhdf5.so.200 (0x00007fc891199000)
libz.so.1 => /lib64/libz.so.1 (0x00007fc890f74000)
libdl.so.2 => /lib64/libdl.so.2 (0x00007fc890d70000)
libgfortran.so.3 => /lib64/libgfortran.so.3 (0x00007fc890a43000)
libm.so.6 => /lib64/libm.so.6 (0x00007fc890741000)
libmpifort.so.12 => /home/gmatteo/local/lib/libmpifort.so.12 (0x00007fc89050a000)
libmpi.so.12 => /home/gmatteo/local/lib/libmpi.so.12 (0x00007fc88ffb9000)
libquadmath.so.0 => /lib64/libquadmath.so.0 (0x00007fc88fd7a000)
libgcc_s.so.1 => /lib64/libgcc_s.so.1 (0x00007fc88fb63000)
libc.so.6 => /lib64/libc.so.6 (0x00007fc88f7a1000)
...
<snip>
As expected, our executable uses the openblas, netcdf, hdf5, mpi libraries installed in $HOME/local/lib plus
other basic libs coming from lib64(e.g. libgfortran) added by the compiler.
Tip
On macOS, replace ldd with otool and the syntax:
otool -L abinit
If you see entries like:
/System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libLAPACK.dylib (compatibility version 1.0.0, current version 1.0.0)
/System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib (compatibility version 1.0.0, current version 1.0.0)
it means that you are linking against macOS VECLIB.
In this case, make sure to use --enable-zdot-bugfix="yes" when configuring the package
otherwise the code will crash at runtime due to ABI incompatibility (calling conventions
for functions returning complex values).
Did I tell you that macOS does not care about Fortran?
If you wonder about the difference between API and ABI, please read this
stackoverflow post.
To understand why LD_LIBRARY_PATH is so important, let’s try to reset the value of this variable with
unset LD_LIBRARY_PATH
echo $LD_LIBRARY_PATH
then rerun ldd (or otool) again.
Do you understand what’s happening here?
Why it’s not possible to execute abinit with an empty $LD_LIBRARY_PATH?
How would you fix the problem?
Troubleshooting¶
Problems can appear at different levels:
- configuration time
- compilation time
- runtime i.e. when executing the code
Configuration-time errors are usually due to misconfiguration of the environment, missing (hard) dependencies or critical problems in the software stack that will make configure abort. Unfortunately, the error message reported by configure is not always self-explanatory. To pinpoint the source of the problem you will need to search for clues in config.log, especially the error messages associated to the feature/library that is triggering the error.
This is not as easy as it looks since configure sometimes performs multiple tests to detect your architecture and some of these tests are supposed to fail. As a consequence, not all the error messages reported in config.log are necessarily relevant. Even if you find the test that makes configure abort, the error message may be obscure and difficult to decipher. In this case, you can ask for help on the forum but remember to provide enough info on your architecture, the compilation options and, most importantly, a copy of config.log. Without this file, indeed, it is almost impossible to understand what’s going on.
An example will help.
Let’s assume we are compiling on a cluster using modules provided by our sysadmin.
More specifically, there is an openmpi_intel2013_sp1.1.106 module that is supposed to provide
the openmpi implementation of the MPI library compiled with a particular version of the intel compiler
(remember what we said about using the same version of the compiler).
Obviously we need to load the modules before running configure in order to setup our environment
so we issue:
module load openmpi_intel2013_sp1.1.106
The module seems to work as no error message is printed to the terminal and which mpicc shows
that the compiler has been added to $PATH.
At this point we try to configure ABINIT with:
with_mpi_prefix="${MPI_HOME}"
where $MPI_HOME is a environment variable set by module load (use e.g. env | grep MPI).
Unfortunately, the configure script aborts at the very beginning complaining
that the C compiler does not work!
checking for gcc... /cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/bin/mpicc
checking for C compiler default output file name...
configure: error: in `/home/gmatteo/abinit/build':
configure: error: C compiler cannot create executables
See `config.log' for more details.
Let’s analyze the output of configure. The line:
checking for gcc... /cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/bin/mpicc
indicates that configure was able to find mpicc in ${MPI_HOME}/bin. Then an internal test is executed to make sure the wrapper can compile a rather simple Fortran program using MPI but the test fails and configure aborts immediately with the pretty explanatory message:
configure: error: C compiler cannot create executables
See `config.log' for more details.
If we want to understand why configure failed, we have to open config.log in the editor and search for error messages towards the end of the log file. For example one can search for the string “C compiler cannot create executables”. Immediately above this line, we find the following section:
config.log
configure:12104: checking whether the C compiler works
configure:12126: /cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/bin/mpicc conftest.c >&5
/cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/lib/libmpi.so: undefined reference to `ibv_reg_xrc_rcv_qp@IBVERBS_1.1'
/cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/lib/libmpi.so: undefined reference to `ibv_modify_xrc_rcv_qp@IBVERBS_1.1'
/cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/lib/libmpi.so: undefined reference to `ibv_open_xrc_domain@IBVERBS_1.1'
/cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/lib/libmpi.so: undefined reference to `ibv_unreg_xrc_rcv_qp@IBVERBS_1.1'
/cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/lib/libmpi.so: undefined reference to `ibv_query_xrc_rcv_qp@IBVERBS_1.1'
/cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/lib/libmpi.so: undefined reference to `ibv_create_xrc_rcv_qp@IBVERBS_1.1'
/cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/lib/libmpi.so: undefined reference to `ibv_create_xrc_srq@IBVERBS_1.1'
/cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/lib/libmpi.so: undefined reference to `ibv_close_xrc_domain@IBVERBS_1.1'
configure:12130: $? = 1
configure:12168: result: no
configure: failed program was:
| /* confdefs.h */
| #define PACKAGE_NAME "ABINIT"
| #define PACKAGE_TARNAME "abinit"
| #define PACKAGE_VERSION "9.1.2"
| #define PACKAGE_STRING "ABINIT 9.1.2"
| #define PACKAGE_BUGREPORT "https://bugs.launchpad.net/abinit/"
| #define PACKAGE_URL ""
| #define PACKAGE "abinit"
| #define VERSION "9.1.2"
| #define ABINIT_VERSION "9.1.2"
| #define ABINIT_VERSION_MAJOR "9"
| #define ABINIT_VERSION_MINOR "1"
| #define ABINIT_VERSION_MICRO "2"
| #define ABINIT_VERSION_BUILD "20200824"
| #define ABINIT_VERSION_BASE "9.1"
| #define HAVE_OS_LINUX 1
| /* end confdefs.h. */
|
| int
| main ()
| {
|
| ;
| return 0;
| }
The line
configure:12126: /cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/bin/mpicc conftest.c >&5
tells us that configure tried to compile a C file named conftest.c and that the return value
stored in the $? shell variable is non-zero thus indicating failure:
configure:12130: $? = 1
configure:12168: result: no
The failing program (the C main after the line “configure: failed program was:”) is a rather simple piece of code and our mpicc compiler is not able to compile it! If we look more carefully at the lines after the invocation of mpicc, we see lots of undefined references to functions of the libibverbs library:
configure:12126: /cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/bin/mpicc conftest.c >&5
/cm/shared/apps/openmpi/1.7.5/intel2013_sp1.1.106/lib/libmpi.so: undefined reference to `ibv_reg_xrc_rcv_qp@IBVERBS_1.1
This looks like some mess in the system configuration and not necessarily a problem in the ABINIT build system. Perhaps there have been changes to the environment, maybe a system upgrade or the module is simply broken. In this case you should send the config.log to the sysadmin so that he/she can fix the problem or just use another more recent module.
Obviously, one can encounter cases in which modules are properly configured yet the configure script aborts because it does not know how to deal with your software stack. In both cases, config.log is key to pinpoint the problem and sometimes you will find that the problem is rather simple to solve. For instance, you are using a Fortran module files produced by gfortran while trying to compile with the intel compiler or perhaps you are trying to use modules produced by a different version of the same compiler. Perhaps you forgot to add the include directory required by an external library and the compiler cannot find the include file or maybe there is a typo in the configuration options. The take-home message is that several mistakes can be detected by just inspecting the log messages reported in configure.log if you know how to search for them.
Compilation-time errors are usually due to syntax errors, portability issues or
Fortran constructs that are not supported by that particular version of the compiler.
In the first two cases, please report the problem on the forum.
In the later case, you will need a more recent version of the compiler.
Sometimes the compilation aborts with an internal compiler error that should be considered
as a bug in the compiler rather than an error in the ABINIT source code.
Decreasing the optimization level when compiling the particular routine that triggers the error
(use -O1 or even -O0 for the most problematic cases) may solve the problem else
try a more recent version of the compiler.
If you have made non-trivial changes in code (modifications in the datatypes/interfaces),
run make clean and recompile.
Runtime errors are more difficult to fix as they may require the use of a debugger and some basic understanding of Linux signals. Here we focus on two common scenarios: SIGILL and SIGSEGV.
If the code raises the SIGILL signal, it means that the CPU attempted to execute an instruction it didn’t understand. Very likely, your executables/libraries have been compiled for the wrong architecture. This may happen on clusters when the CPU family available on the frontend differs from the one available on the compute node and aggressive optimization options (-O3, -march, -xHost, etc) are used. Removing the optimization options and using the much safer -O2 level may help. Alternatively, one can configure and compile the source directly on the compute node or use compilation options compatible both with the frontend and the compute node (ask your sysadmin for details).
Warning
Never ever run calculations on CPUs belonging to different families unless you know what you are doing. Many MPI codes assume reproducibility at the binary level: on different MPI processes the same set of bits in input should produce the same set of bits in output. If you are running on a heterogeneous cluster, select the queue with the same CPU family and make sure the code has been compiled with options that are compatibile with the compute node.
Segmentation faults (SIGSEGV) are usually due to bugs in the code but they may also be triggered by non-portable code or misconfiguration of the software stack. When reporting this kind of problem on the forum, please add an input file so that developers can try to reproduce the problem. Keep in mind, however, that the problem may not be reproducible on other architectures. The ideal solution would be to run the code under the control of the debugger, use the backtrace to locate the line of code where the segmentation fault occurs and then attach the backtrace to your issue on the forum.
How to run gdb
Using the debugger in sequential is really simple.
First of all, make sure the code have been compiled with the -g option
to generate source-level debug information.
To use the gdb GNU debugger, perform the following operations:
-
Load the executable in the GNU debugger using the syntax:
gdb path_to_abinit_executable -
Run the code with the
runcommand and pass the input file as argument:(gdb) run t02.in -
Wait for the error e.g. SIGSEGV, then print the backtrace with:
(gdb) bt
PS: avoid debugging code compiled with -O3 or -Ofast as the backtrace may not be reliable.
Sometimes, even -O2 (default) is not reliable and you have to resort to print statements
and bisection to braket the problematic piece of code.
How to compile ABINIT on a cluster with the intel toolchain and modules¶
On intel-based clusters, we suggest to compile ABINIT with the intel compilers (icc and ifort) and MKL in order to achieve better performance. The MKL library, indeed, provides highly-optimized implementations for BLAS, LAPACK, FFT, and SCALAPACK that can lead to a significant speedup while simplifying considerably the compilation process. As concerns MPI, intel provides its own implementation (Intel MPI) but it is also possible to employ openmpi or mpich provided these libraries have been compiled with the same intel compilers.
In what follows, we assume a cluster in which scientific software is managed with modules and the EasyBuild framework. Before proceeding with the next steps, it is worth summarizing the most important module commands.
module commands
To list the modules installed on the cluster, use:
module avail
The syntax to load the module MODULE_NAME is:
module load MODULE_NAME
while
module list
prints the list of modules currently loaded.
To list all modules containing “string”, use:
module spider string # requires LMOD with LUA
Finally,
module show MODULE_NAME
shows the commands in the module file (useful for debugging). For a more complete introduction to environment modules, please consult this page.
On my cluster, I can activate intel MPI by executing:
module load releases/2018b
module load intel/2018b
module load iimpi/2018b
to load the 2018b intel MPI EasyBuild toolchain. On your cluster, you may need to load different modules but the effect at the level of the shell environment should be the same. More specifically, mpiifort is now in PATH (note how mpiifort wraps intel ifort):
mpiifort -v
mpiifort for the Intel(R) MPI Library 2018 Update 3 for Linux*
Copyright(C) 2003-2018, Intel Corporation. All rights reserved.
ifort version 18.0.3
the directories with the libraries required by the compiler/MPI have been added
to LD_LIBRARY_PATH while CPATH stores the locations to search for include file.
Last but not least, the env should now define intel-specific variables
whose name starts with I_:
$ env | grep I_
I_MPI_ROOT=/opt/cecisw/arch/easybuild/2018b/software/impi/2018.3.222-iccifort-2018.3.222-GCC-7.3.0-2.30
Since I_MPI_ROOT points to the installation directory of intel MPI, we can use this environment variable to tell configure how to locate our MPI installation:
with_mpi="${I_MPI_ROOT}"
FC="mpiifort" # Use intel wrappers. Important!
CC="mpiicc" # See warning below
CXX="mpiicpc"
# with_optim_flavor="aggressive"
# FCFLAGS="-g -O2"
Optionally, you can use with_optim_flavor="aggressive to let configure select compilations
options tuned for performance or set the options explicitly via FCFLAGS.
Warning
Intel MPI installs two sets of MPI wrappers.
(mpiicc, mpicpc, mpiifort) and (mpicc, mpicxx, mpif90) that use
Intel compilers and GNU compilers, respectively.
Use the -show option (e.g. mpif90 -show) to display the underlying compiler.
As expected
$ mpif90 -v
mpif90 for the Intel(R) MPI Library 2018 Update 3 for Linux*
COLLECT_GCC=gfortran
<snip>
Thread model: posix
gcc version 7.3.0 (GCC)
shows that mpif90 wraps GNU gfortran.
Unless you really need to use GNU compilers, we strongly suggest the wrappers
based on the Intel compilers (mpiicc, mpicpc, mpiifort)
If we run configure with these options, we should see a section at the beginning in which the build system is testing basic capabilities of the Fortran compiler. If configure stops at this level it means there’s a severe problem with your toolchain.
==============================================================================
=== Fortran support ===
==============================================================================
checking for mpiifort... /opt/cecisw/arch/easybuild/2018b/software/impi/2018.3.222-iccifort-2018.3.222-GCC-7.3.0-2.30/bin64/mpiifort
checking whether we are using the GNU Fortran compiler... no
checking whether mpiifort accepts -g... yes
checking which type of Fortran compiler we have... intel 18.0
Then we have a section in which configure tests the MPI implementation:
Multicore architecture support
==============================================================================
=== Multicore architecture support ===
==============================================================================
checking whether to enable OpenMP support... no
checking whether to enable MPI... yes
checking how MPI parameters have been set... yon
checking whether the MPI C compiler is set... yes
checking whether the MPI C++ compiler is set... yes
checking whether the MPI Fortran compiler is set... yes
checking for MPI C preprocessing flags...
checking for MPI C flags...
checking for MPI C++ flags...
checking for MPI Fortran flags...
checking for MPI linker flags...
checking for MPI library flags...
checking whether the MPI C API works... yes
checking whether the MPI C environment works... yes
checking whether the MPI C++ API works... yes
checking whether the MPI C++ environment works... yes
checking whether the MPI Fortran API works... yes
checking whether the MPI Fortran environment works... yes
checking whether to build MPI I/O code... auto
checking which level of MPI is supported by the Fortran compiler... 2
configure: forcing MPI-2 standard level support
checking whether the MPI library supports MPI_INTEGER16... yes
checking whether the MPI library supports MPI_CREATE_TYPE_STRUCT... yes
checking whether the MPI library supports MPI_IBCAST (MPI3)... yes
checking whether the MPI library supports MPI_IALLGATHER (MPI3)... yes
checking whether the MPI library supports MPI_IALLTOALL (MPI3)... yes
checking whether the MPI library supports MPI_IALLTOALLV (MPI3)... yes
checking whether the MPI library supports MPI_IGATHERV (MPI3)... yes
checking whether the MPI library supports MPI_IALLREDUCE (MPI3)... yes
configure:
configure: dumping all MPI parameters for diagnostics
configure: ------------------------------------------
configure:
configure: Configure options:
configure:
configure: * enable_mpi_inplace = ''
configure: * enable_mpi_io = ''
configure: * with_mpi = 'yes'
configure: * with_mpi_level = ''
configure:
configure: Internal parameters
configure:
configure: * MPI enabled (required) : yes
configure: * MPI C compiler is set (required) : yes
configure: * MPI C compiler works (required) : yes
configure: * MPI Fortran compiler is set (required) : yes
configure: * MPI Fortran compiler works (required) : yes
configure: * MPI environment usable (required) : yes
configure: * MPI C++ compiler is set (optional) : yes
configure: * MPI C++ compiler works (optional) : yes
configure: * MPI-in-place enabled (optional) : no
configure: * MPI-IO enabled (optional) : yes
configure: * MPI configuration type (computed) : yon
configure: * MPI Fortran level supported (detected) : 2
configure: * MPI_Get_library_version available (detected) : unknown
configure:
configure: All required parameters must be set to 'yes'.
configure: If not, the configuration and/or the build with
configure: MPI support will very likely fail.
configure:
checking whether to activate GPU support... no
So far so good. Our compilers and MPI seem to work so we can proceed with the setup of the external libraries.
On my cluster, module load intel/2018b has also defined the MKLROOT env variable
env | grep MKL
MKLROOT=/opt/cecisw/arch/easybuild/2018b/software/imkl/2018.3.222-iimpi-2018b/mkl
EBVERSIONIMKL=2018.3.222
that can be used in conjunction with the highly recommended mkl-link-line-advisor to link with MKL. On other clusters, you may need load an mkl module explicitly (or composerxe or parallel-studio-xe)
Let’s now discuss how to configure ABINIT with MKL starting from the simplest cases:
- BLAS and Lapack from MKL
- FFT from MKL DFTI
- no Scalapack
- no OpenMP threads.
These are the options I have to select in the mkl-link-line-advisor to enable this configuration with my software stack:
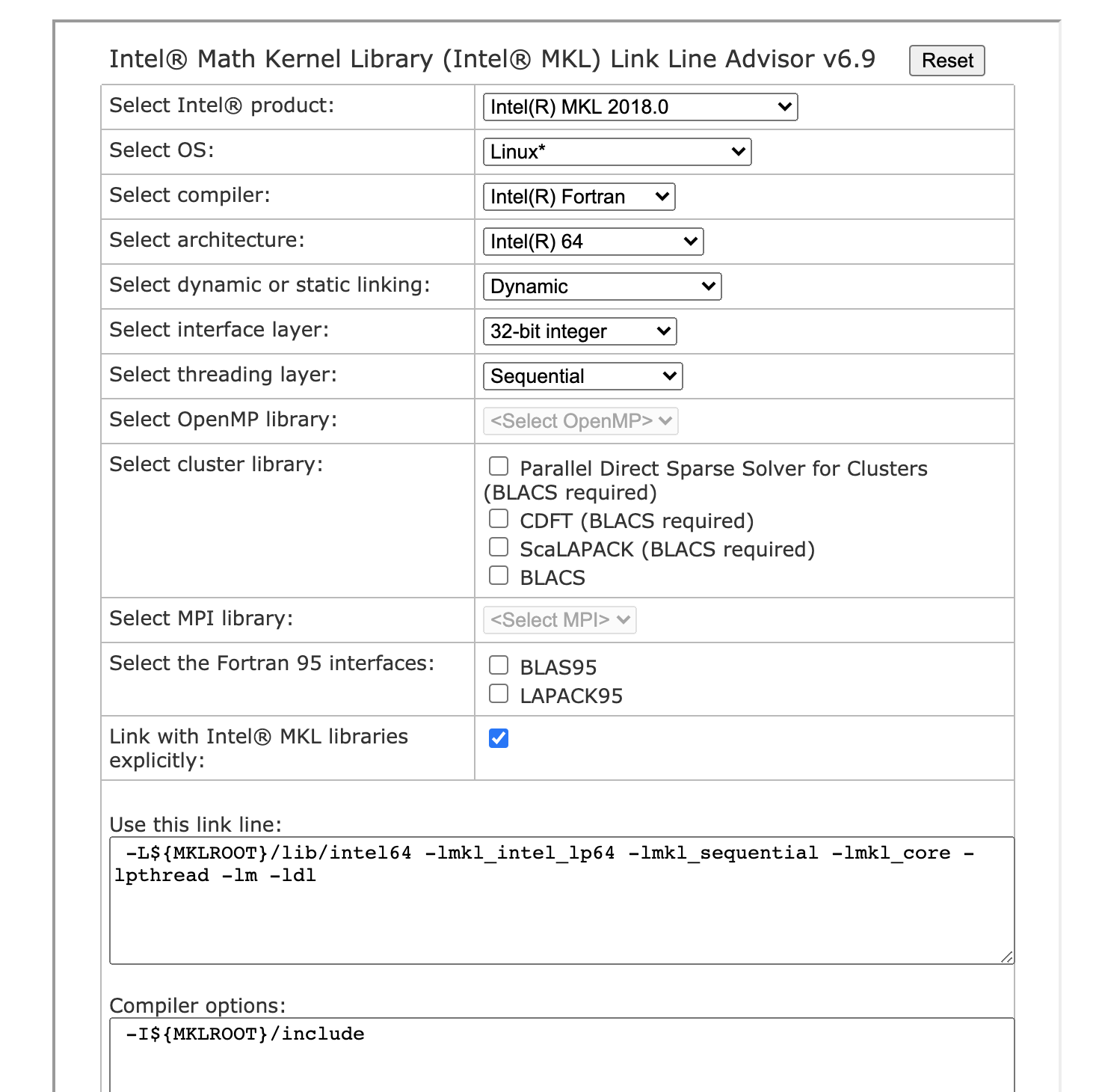
The options should be self-explanatory. Perhaps the tricky part is Select interface layer where one should select 32-bit integer. This simply means that we are compiling and linking code in which default integer is 32-bits wide (default behaviour). Note how the threading layer is set to Sequential (no OpenMP threads) and how we chose to link with MKL libraries explicitly the get the full link line and compiler options.
Now we can use these options in our configuration file:
# BLAS/LAPACK with MKL
with_linalg_flavor="mkl"
LINALG_CPPFLAGS="-I${MKLROOT}/include"
LINALG_FCFLAGS="-I${MKLROOT}/include"
LINALG_LIBS="-L${MKLROOT}/lib/intel64 -lmkl_intel_lp64 -lmkl_core -lmkl_sequential -lpthread -lm -ldl"
# FFT from MKL
with_fft_flavor="dfti"
FFT_CPPFLAGS="-I${MKLROOT}/include"
FFT_FCFLAGS="-I${MKLROOT}/include"
FFT_LIBS="-L${MKLROOT}/lib/intel64 -lmkl_intel_lp64 -lmkl_core -lmkl_sequential -lpthread -lm -ldl"
Warning
Do not use MKL with FFTW3 for the FFT as the MKL library exports the same symbols as FFTW. This means that the linker will receive multiple definitions for the same procedure and the behaviour is undefined! Use either MKL or FFTW3 with e.g. openblas.
If we run configure with these options, we should obtain the following output in the Linear algebra support section:
Linear algebra support
==============================================================================
=== Linear algebra support ===
==============================================================================
checking for the requested linear algebra flavor... mkl
checking for the serial linear algebra detection sequence... mkl
checking for the MPI linear algebra detection sequence... mkl
checking for the MPI acceleration linear algebra detection sequence... none
checking how to detect linear algebra libraries... verify
checking for BLAS support in the specified libraries... yes
checking for AXPBY support in the BLAS libraries... yes
checking for GEMM3M in the BLAS libraries... yes
checking for mkl_imatcopy in the specified libraries... yes
checking for mkl_omatcopy in the specified libraries... yes
checking for mkl_omatadd in the specified libraries... yes
checking for mkl_set/get_threads in the specified libraries... yes
checking for LAPACK support in the specified libraries... yes
checking for LAPACKE C API support in the specified libraries... no
checking for PLASMA support in the specified libraries... no
checking for BLACS support in the specified libraries... no
checking for ELPA support in the specified libraries... no
checking how linear algebra parameters have been set... env (flavor: kwd)
checking for the actual linear algebra flavor... mkl
checking for linear algebra C preprocessing flags... none
checking for linear algebra C flags... none
checking for linear algebra C++ flags... none
checking for linear algebra Fortran flags... -I/opt/cecisw/arch/easybuild/2018b/software/imkl/2018.3.222-iimpi-2018b/mkl/include
checking for linear algebra linker flags... none
checking for linear algebra library flags... -L/opt/cecisw/arch/easybuild/2018b/software/imkl/2018.3.222-iimpi-2018b/mkl/lib/intel64 -lmkl_intel_lp64 -lmkl_core -lmkl_sequential -lpthread -lm -ldl
configure: WARNING: parallel linear algebra is not available
Excellent, configure detected a working BLAS/Lapack installation, plus some MKL extensions (mkl_imatcopy etc). BLACS and Scalapack (parallel linear algebra) have not been detected but this is expected as we haven’t asked for these libraries in the mkl-link-line-advisor GUI.
This is the section in which configure checks the presence of the FFT library (DFTI from MKL, goedecker means internal Fortran version).
Optimized FFT support
==============================================================================
=== Optimized FFT support ===
==============================================================================
checking which FFT flavors to enable... dfti goedecker
checking for FFT flavor... dfti
checking for FFT C preprocessing flags...
checking for FFT C flags...
checking for FFT Fortran flags...
checking for FFT linker flags...
checking for FFT library flags...
checking for the FFT flavor to try... dfti
checking whether to enable DFTI... yes
checking how DFTI parameters have been set... mkl
checking for DFTI C preprocessing flags... none
checking for DFTI C flags... none
checking for DFTI Fortran flags... -I/opt/cecisw/arch/easybuild/2018b/software/imkl/2018.3.222-iimpi-2018b/mkl/include
checking for DFTI linker flags... none
checking for DFTI library flags... -L/opt/cecisw/arch/easybuild/2018b/software/imkl/2018.3.222-iimpi-2018b/mkl/lib/intel64 -lmkl_intel_lp64 -lmkl_core -lmkl_sequential -lpthread -lm -ldl
checking whether the DFTI library works... yes
checking for the actual FFT flavor to use... dfti
The line
checking whether the DFTI library works... yes
tells us that DFTI has been found and we can link against it although this does not necessarily mean that the final executable will work out of the box.
Tip
You may have noticed that it is also possible to use MKL with GNU gfortran but in this case you need to use a different set of libraries including the so-called compatibility layer that allows GCC code to call MKL routines. Also, MKL Scalapack requires either Intel MPI or MPICH2.
Optional Exercise
Compile ABINIT with BLAS/ScalaPack from MKL. Scalapack (or ELPA) may lead to a significant speedup when running GS calculations with large nband. See also the np_slk input variable.
How to compile libxc, netcdf4/hdf5 with intel¶
At this point, one should check whether our cluster provides modules for
libxc, netcdf-fortran, netcdf-c and hdf5 compiled with the same toolchain.
Use module spider netcdf or module keyword netcdf to find the modules (if any).
Hopefully, you will find a pre-existent installation for netcdf and hdf5 (possibly with MPI-IO support)
as these libraries are quite common on HPC centers.
Load these modules to have nc-config and nf-config in your $PATH and then use the
--prefix option to specify the installation directory as done in the previous examples.
Unfortunately, libxc and hdf5 do not provide similar tools so you will have to find
the installation directory for these libs and pass it to configure.
Tip
You may encounter problems with libxc as this library is rather domain-specific and not all the HPC centers install it. If your cluster does not provide libxc, it should not be that difficult to reuse the expertise acquired in this tutorial to build your version and then install the missing dependencies inside $HOME/local. Just remember to:
- load the correct modules for MPI with the associated compilers before configuring
- configure with CC=mpiicc and FC=mpiifort so that the intel compilers are used
- install the libraries and prepend $HOME/local/lib to LD_LIBRARY_PATH
- use the with_LIBNAME option in conjunction with $HOME/local/lib in the ac9 file.
- run configure with the ac9 file.
In the worst case scenario in which neither netcdf4/hdf5 nor libxc are installed, you may want to use the internal fallbacks. The procedure goes as follows.
- Start to configure with a minimalistic set of options just for MPI and MKL (linalg and FFT)
- The build system will detect that some hard dependencies are missing and will generate a build-abinit-fallbacks.sh script in the fallbacks directory.
- Execute the script to build the missing dependencies using the toolchain specified in the initial configuration file
- Finally, reconfigure ABINIT with the fallbacks.
How to compile ABINIT with support for OpenMP threads¶
Tip
For a quick introduction to MPI and OpenMP and a comparison between the two parallel programming paradigms, see this presentation.
Compiling ABINIT with OpenMP is not that difficult as everything boils down to:
- Using a threaded version for BLAS, LAPACK and FFTs
- Passing enable_openmp=”yes” to the ABINIT configure script so that OpenMP is activated also at level of the ABINIT Fortran code.
On the contrary, answering the questions:
- When and why should I use OpenMP threads for my calculations?
- How many threads should I use and what is the parallel speedup I should expect?
is much more difficult as there are several factors that should be taken into account.
Note
To keep a long story short, one should use OpenMP threads when we start to trigger limitations or bottlenecks in the MPI implementation, especially at the level of the memory requirements or in terms of parallel scalability. These problems are usually observed in calculations with large natom, %mpw, nband.
As a matter of fact, it does not make sense to compile ABINIT with OpenMP if your calculations are relatively small. Indeed, ABINIT is mainly designed with MPI-parallelism in mind. For instance, calculations done with a relatively large number of \(\kk\)-points will benefit more of MPI than OpenMP, especially if the number of MPI processes divides the number of \(\kk\)-points exactly. Even worse, do not compile the code with OpenMP support if you do not plan to use threads because the OpenMP version will have an additional overhead due to the creation of the threaded sections.
Remember also that increasing the number of threads does not necessarily leads to faster calculations (the same is true for MPI processes). There’s always an optimal value for the number of threads (MPI processes) beyond which the parallel efficiency starts to deteriorate. Unfortunately, this value is strongly hardware and software dependent so you will need to benchmark the code before running production calculations.
Last but not least, OpenMP threads are not necessarily Posix threads. Hence if you have a library that provides both Open and Posix-threads, link with the OpenMP version.
After this necessary preamble, let’s discuss how to compile a threaded version. To activate OpenMP support in the Fortran routines of ABINIT, pass
enable_openmp="yes"
to the configure script via the configuration file.
This will automatically activate the compilation option needed to enable OpenMP in the ABINIT source code
(e..g. -fopenmp option for gfortran) and the CPP variable HAVE_OPENMP in config.h.
Note that this option is just part of the story as a significant fraction of the wall-time is spent in the external
BLAS/FFT routines so do not expect big speedups if you do not link against threaded libraries.
If you are building your own software stack for BLAS/LAPACK and FFT, you will have to reconfigure with the correct options for the OpenMP version and then issue make and make install again to build the threaded version. Also note that some libraries may change. FFTW3, for example, ships the OpenMP version in libfftw3_omp (see the official documentation) hence the list of libraries in FFTW3_LIBS should be changed accordingly.
Life is much easier if you are using intel MKL because in this case
it is just a matter of selecting OpenMP threading as threading layer
in the mkl-link-line-advisor interface
and then pass these options to the ABINIT build system together with enable_openmp="yes".
Important
When using threaded libraries remember to set explicitly the number of threads with e.g.
export OMP_NUM_THREADS=2
either in your bash_profile or in the submission script (or in both). By default, OpenMP uses all the available CPUs so it is very easy to overload the machine, especially if one uses threads in conjunction with MPI processes.
When running threaded applications with MPI, we suggest to allocate a number of physical CPUs that is equal to the number of MPI processes times the number of OpenMP threads. Computational intensive applications such as DFT codes have less chance to be improved in performance from Hyper-Threading technology (usually referred to as number of logical CPUs).
We also recommend to increase the stack size limit using e.g.
ulimit -s unlimited
if the sysadmin allows you to do so.
To run the ABINIT test suite with e.g. two OpenMP threads, use the -o2 option of runtests.py