Fourth tutorial¶
Aluminum, the bulk and the surface.¶
This tutorial aims at showing how to get the following physical properties for a metal and for a surface:
- the total energy
- the lattice parameter
- the relaxation of surface atoms
- the surface energy
You will learn about the smearing of the Brillouin zone integration, and also a bit about preconditioning the SCF cycle.
This tutorial should take about 1 hour and 30 minutes.
Note
Supposing you made your own installation of ABINIT, the input files to run the examples are in the ~abinit/tests/ directory where ~abinit is the absolute path of the abinit top-level directory. If you have NOT made your own install, ask your system administrator where to find the package, especially the executable and test files.
In case you work on your own PC or workstation, to make things easier, we suggest you define some handy environment variables by executing the following lines in the terminal:
export ABI_HOME=Replace_with_absolute_path_to_abinit_top_level_dir # Change this line
export PATH=$ABI_HOME/src/98_main/:$PATH # Do not change this line: path to executable
export ABI_TESTS=$ABI_HOME/tests/ # Do not change this line: path to tests dir
export ABI_PSPDIR=$ABI_TESTS/Pspdir/ # Do not change this line: path to pseudos dir
Examples in this tutorial use these shell variables: copy and paste
the code snippets into the terminal (remember to set ABI_HOME first!) or, alternatively,
source the set_abienv.sh script located in the ~abinit directory:
source ~abinit/set_abienv.sh
The ‘export PATH’ line adds the directory containing the executables to your PATH so that you can invoke the code by simply typing abinit in the terminal instead of providing the absolute path.
To execute the tutorials, create a working directory (Work*) and
copy there the input files of the lesson.
Most of the tutorials do not rely on parallelism (except specific tutorials on parallelism). However you can run most of the tutorial examples in parallel with MPI, see the topic on parallelism.
Total energy and lattice parameters at fixed smearing and k-point grid¶
Before beginning, you might consider to work in a different subdirectory, as for tutorials 1, 2 or 3. Why not Work4?
The following commands will move you to your working directory, create the Work4 directory, and move you into that directory as you did in the previous tutorials. Then, we copy the file tbase4_1.abi inside the Work4 directory. The commands are:
cd $ABI_TESTS/tutorial/Input
mkdir Work4
cd Work4
cp ../tbase4_1.abi .
tbase4_1.abi is our input file. You should edit it and read it carefully,
# Crystalline aluminum : optimization of the lattice parameter # at fixed number of k points and broadening. #Definition of the unit cell acell 3*7.60 # This is equivalent to 7.60 7.60 7.60 rprim 0.0 0.5 0.5 # FCC primitive vectors (to be scaled by acell) 0.5 0.0 0.5 0.5 0.5 0.0 #Definition of the atom types ntypat 1 # There is only one type of atom znucl 13 # The keyword "znucl" refers to the atomic number of the # possible type(s) of atom. The pseudopotential(s) # mentioned in the "files" file must correspond # to the type(s) of atom. Here, the only type is Aluminum pp_dirpath "$ABI_PSPDIR" # This is the path to the directory were # pseudopotentials for tests are stored pseudos "Psdj_nc_sr_04_pw_std_psp8/Al.psp8" # Name and location of the pseudopotential #Definition of the atoms natom 1 # There is only one atom per cell typat 1 # This atom is of type 1, that is, Aluminum xred # This keyword indicate that the location of the atoms # will follow, one triplet of number for each atom 0.0 0.0 0.0 # Triplet giving the REDUCED coordinate of atom 1. #Definition of the planewave basis set ecut 6.0 # Maximal kinetic energy cut-off, in Hartree #Definition of the k-point grid ngkpt 2 2 2 # This is a 2x2x2 FCC grid, based on the primitive vectors nshiftk 4 # of the reciprocal space. For a FCC real space lattice, # like the present one, it actually corresponds to the # so-called 4x4x4 Monkhorst-Pack grid, if the following shifts # are used : shiftk 0.5 0.5 0.5 0.5 0.0 0.0 0.0 0.5 0.0 0.0 0.0 0.5 #Definition of the SCF procedure nstep 10 # Maximal number of SCF cycles toldfe 1.0d-6 # Will stop when, twice in a row, the difference # between two consecutive evaluations of total energy # differ by less than toldfe (in Hartree) # This value is WAY TOO LARGE for most realistic studies of materials #Definition of occupation numbers occopt 4 tsmear 0.05 #Optimization of the lattice parameters optcell 1 geoopt "bfgs" ntime 10 dilatmx 1.05 ecutsm 0.5 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% tbase4_1.abo, tolnlines= 6, tolabs= 1.2e-07, tolrel= 1.2e-03 #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = Unknown #%% keywords = #%% description = #%% Crystalline aluminum : optimization of the lattice parameter #%% at fixed number of k points and broadening. #%%<END TEST_INFO>
and then a look at the following new input variables:
Note also the following:
-
We will work at fixed ecut (6Ha). It is implicit that in real research application, you should do a convergence test with respect to ecut. Here, a suitable ecut is given to you in order to save time. It will give a lattice parameter that is 0.2% off of the experimental value. Note that this is the softest pseudopotential of those that we have used until now: the 01h.pspgth for H needed 30 Ha (it was rather hard), the Si.psp8 for Si needed 12 Ha. See the end of this page for a discussion of soft and hard pseudopotentials.
-
The input variable diemac has been suppressed. Aluminum is a metal, and the default value for this input variable is tailored for that case.
When you have read the input file, you can run the code, as usual (it will take a few seconds).
abinit tbase4_1.abi > log 2> err &
Then, give a quick look at the output file. You should note that the Fermi energy and occupation numbers have been computed automatically:
Fermi (or HOMO) energy (hartree) = 0.27151 Average Vxc (hartree)= -0.36713
Eigenvalues (hartree) for nkpt= 2 k points:
kpt# 1, nband= 3, wtk= 0.75000, kpt= -0.2500 0.5000 0.0000 (reduced coord)
0.09836 0.25743 0.42131
occupation numbers for kpt# 1
2.00003 1.33305 0.00015
prteigrs : prtvol=0 or 1, do not print more k-points.
You should also note that the components of the total energy include an entropy term:
--- !EnergyTerms
iteration_state : {dtset: 1, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.68009594268178E-01
hartree : 3.75144741427686E-03
xc : -1.11506134985146E+00
Ewald energy : -2.71387012800927E+00
psp_core : 1.56870175692757E-02
local_psp : 1.66222476058238E-01
non_local_psp : 4.25215770913582E-01
internal : -2.35004517163717E+00
'-kT*entropy' : -7.99850001032776E-03
total_energy : -2.35804367164750E+00
total_energy_eV : -6.41656315078440E+01
band_energy : 3.72511439902163E-01
...
The convergence study with respect to k-points¶
There is of course a convergence study associated to the sampling of the Brillouin zone. You should examine different grids, of increasing resolution. You might try the following series of grids:
ngkpt1 2 2 2
ngkpt2 4 4 4
ngkpt3 6 6 6
ngkpt4 8 8 8
with the associated nkpt:
nkpt1 2
nkpt2 10
nkpt3 28
nkpt4 60
The input file tbase4_2.abi is an example:
# Crystalline aluminum : optimization of the lattice parameter # # Convergence with respect to k points. ndtset 4 getwfk -1 #Definition of the unit cell acell 3*7.60 # This is equivalent to 7.60 7.60 7.60 rprim 0.0 0.5 0.5 # FCC primitive vectors (to be scaled by acell) 0.5 0.0 0.5 0.5 0.5 0.0 #Definition of the atom types ntypat 1 # There is only one type of atom znucl 13 # The keyword "znucl" refers to the atomic number of the # possible type(s) of atom. The pseudopotential(s) # mentioned in the "files" file must correspond # to the type(s) of atom. Here, the only type is Aluminum pp_dirpath "$ABI_PSPDIR" # This is the path to the directory were # pseudopotentials for tests are stored pseudos "Psdj_nc_sr_04_pw_std_psp8/Al.psp8" # Name and location of the pseudopotential #Definition of the atoms natom 1 # There is only one atom per cell typat 1 # This atom is of type 1, that is, Aluminum xred # This keyword indicate that the location of the atoms # will follow, one triplet of number for each atom 0.0 0.0 0.0 # Triplet giving the REDUCED coordinate of atom 1. #Definition of the planewave basis set ecut 6.0 # Maximal kinetic energy cut-off, in Hartree #Definition of the k-point grids nshiftk 4 shiftk 0.5 0.5 0.5 # These shifts will be the same for all grids 0.5 0.0 0.0 0.0 0.5 0.0 0.0 0.0 0.5 ngkpt1 2 2 2 ngkpt2 4 4 4 ngkpt3 6 6 6 ngkpt4 8 8 8 #Definition of the SCF procedure nstep 10 # Maximal number of SCF cycles tolvrs 1.0d-14 # Will stop when, twice in a row, the difference # between two consecutive evaluations of total energy # differ by less than toldfe (in Hartree) # This value is REASONABLE for most realistic studies of materials #Definition of occupation numbers occopt 4 tsmear 0.05 #Optimization of the lattice parameters optcell 1 geoopt "bfgs" ntime 10 dilatmx 1.05 ecutsm 0.5 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% tbase4_2.abo, tolnlines = 0, tolabs = 0.0e+00, tolrel = 0.0e+00, fld_options = -easy #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = Unknown #%% keywords = #%% description = #%% Crystalline aluminum : optimization of the lattice parameter #%% #%% Convergence with respect to k points. #%%<END TEST_INFO>
while tbase4_2.abo is a reference output file:
.Version 10.5.8.2 of ABINIT, released Oct 2025.
.(MPI version, prepared for a x86_64_linux_gnu13.2 computer)
.Copyright (C) 1998-2026 ABINIT group .
ABINIT comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Sat 20 Dec 2025.
- ( at 17h05 )
- input file -> /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/TestBot_MPI1/tutorial_tbase4_2/tbase4_2.abi
- output file -> tbase4_2.abo
- root for input files -> tbase4_2i
- root for output files -> tbase4_2o
DATASET 1 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 1.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 2
mpw = 90 nfft = 3375 nkpt = 2
================================================================================
P This job should need less than 2.510 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.010 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 2 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 2.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 10
mpw = 92 nfft = 3375 nkpt = 10
================================================================================
P This job should need less than 2.556 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.044 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 3 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 3.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 28
mpw = 94 nfft = 3375 nkpt = 28
================================================================================
P This job should need less than 2.661 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.122 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 4 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 4.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 60
mpw = 95 nfft = 3375 nkpt = 60
================================================================================
P This job should need less than 2.850 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.263 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
--------------------------------------------------------------------------------
------------- Echo of variables that govern the present computation ------------
--------------------------------------------------------------------------------
-
- outvars: echo of selected default values
- iomode0 = 0 , fftalg0 =512 , wfoptalg0 = 0
-
- outvars: echo of global parameters not present in the input file
- max_nthreads = 0
-
-outvars: echo values of preprocessed input variables --------
acell 7.6000000000E+00 7.6000000000E+00 7.6000000000E+00 Bohr
amu 2.69815390E+01
dilatmx 1.05000000E+00
ecut 6.00000000E+00 Hartree
ecutsm 5.00000000E-01 Hartree
- fftalg 512
getwfk -1
ionmov 2
ixc -1012
jdtset 1 2 3 4
kpt1 -2.50000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
kpt2 -1.25000000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.75000000E-01 0.00000000E+00
-3.75000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 1.25000000E-01
-1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
kpt3 -8.33333333E-02 -1.66666667E-01 0.00000000E+00
-8.33333333E-02 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -2.50000000E-01 0.00000000E+00
-8.33333333E-02 -2.50000000E-01 8.33333333E-02
-8.33333333E-02 5.00000000E-01 0.00000000E+00
-1.66666667E-01 -4.16666667E-01 0.00000000E+00
-8.33333333E-02 -4.16666667E-01 8.33333333E-02
-2.50000000E-01 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -3.33333333E-01 8.33333333E-02
-8.33333333E-02 -3.33333333E-01 1.66666667E-01
-8.33333333E-02 3.33333333E-01 0.00000000E+00
-1.66666667E-01 4.16666667E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 0.00000000E+00
-1.66666667E-01 5.00000000E-01 8.33333333E-02
-3.33333333E-01 -4.16666667E-01 0.00000000E+00
-2.50000000E-01 -4.16666667E-01 8.33333333E-02
-1.66666667E-01 -4.16666667E-01 1.66666667E-01
-8.33333333E-02 -4.16666667E-01 2.50000000E-01
-8.33333333E-02 1.66666667E-01 0.00000000E+00
-1.66666667E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.33333333E-01 0.00000000E+00
-3.33333333E-01 4.16666667E-01 0.00000000E+00
-4.16666667E-01 5.00000000E-01 0.00000000E+00
-3.33333333E-01 5.00000000E-01 8.33333333E-02
-2.50000000E-01 5.00000000E-01 1.66666667E-01
-8.33333333E-02 0.00000000E+00 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
-4.16666667E-01 0.00000000E+00 0.00000000E+00
kpt4 -6.25000000E-02 -1.25000000E-01 0.00000000E+00
-6.25000000E-02 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 -1.87500000E-01 0.00000000E+00
-6.25000000E-02 -1.87500000E-01 6.25000000E-02
-6.25000000E-02 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.12500000E-01 0.00000000E+00
-6.25000000E-02 -3.12500000E-01 6.25000000E-02
-1.87500000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 -2.50000000E-01 6.25000000E-02
-6.25000000E-02 -2.50000000E-01 1.25000000E-01
-6.25000000E-02 5.00000000E-01 0.00000000E+00
-1.25000000E-01 -4.37500000E-01 0.00000000E+00
-6.25000000E-02 -4.37500000E-01 6.25000000E-02
-1.87500000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 6.25000000E-02
-6.25000000E-02 -3.75000000E-01 1.25000000E-01
-2.50000000E-01 -3.12500000E-01 0.00000000E+00
-1.87500000E-01 -3.12500000E-01 6.25000000E-02
-1.25000000E-01 -3.12500000E-01 1.25000000E-01
-6.25000000E-02 -3.12500000E-01 1.87500000E-01
-6.25000000E-02 3.75000000E-01 0.00000000E+00
-1.25000000E-01 4.37500000E-01 0.00000000E+00
-1.87500000E-01 5.00000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 6.25000000E-02
-2.50000000E-01 -4.37500000E-01 0.00000000E+00
-1.87500000E-01 -4.37500000E-01 6.25000000E-02
-1.25000000E-01 -4.37500000E-01 1.25000000E-01
-6.25000000E-02 -4.37500000E-01 1.87500000E-01
-3.12500000E-01 -3.75000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 6.25000000E-02
-1.87500000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 -3.75000000E-01 1.87500000E-01
-6.25000000E-02 -3.75000000E-01 2.50000000E-01
-6.25000000E-02 2.50000000E-01 0.00000000E+00
-1.25000000E-01 3.12500000E-01 0.00000000E+00
-1.87500000E-01 3.75000000E-01 0.00000000E+00
-2.50000000E-01 4.37500000E-01 0.00000000E+00
-3.12500000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 6.25000000E-02
-1.87500000E-01 5.00000000E-01 1.25000000E-01
-3.75000000E-01 -4.37500000E-01 0.00000000E+00
-3.12500000E-01 -4.37500000E-01 6.25000000E-02
-2.50000000E-01 -4.37500000E-01 1.25000000E-01
-1.87500000E-01 -4.37500000E-01 1.87500000E-01
-1.25000000E-01 -4.37500000E-01 2.50000000E-01
-6.25000000E-02 -4.37500000E-01 3.12500000E-01
-6.25000000E-02 1.25000000E-01 0.00000000E+00
-1.25000000E-01 1.87500000E-01 0.00000000E+00
-1.87500000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.12500000E-01 0.00000000E+00
outvar_i_n : Printing only first 50 k-points.
kptrlatt1 2 -2 2 -2 2 2 -2 -2 2
kptrlatt2 4 -4 4 -4 4 4 -4 -4 4
kptrlatt3 6 -6 6 -6 6 6 -6 -6 6
kptrlatt4 8 -8 8 -8 8 8 -8 -8 8
kptrlen1 1.52000000E+01
kptrlen2 3.04000000E+01
kptrlen3 4.56000000E+01
kptrlen4 6.08000000E+01
P mkmem1 2
P mkmem2 10
P mkmem3 28
P mkmem4 60
natom 1
nband1 3
nband2 3
nband3 3
nband4 3
ndtset 4
ngfft 15 15 15
nkpt1 2
nkpt2 10
nkpt3 28
nkpt4 60
nstep 10
nsym 48
ntime 10
ntypat 1
occ1 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ2 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ3 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ4 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
prtocc : prtvol=0, do not print more k-points.
occopt 4
optcell 1
rprim 0.0000000000E+00 5.0000000000E-01 5.0000000000E-01
5.0000000000E-01 0.0000000000E+00 5.0000000000E-01
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
shiftk 5.00000000E-01 5.00000000E-01 5.00000000E-01
spgroup 225
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
tolvrs 1.00000000E-14
tsmear 5.00000000E-02 Hartree
typat 1
wtk1 0.75000 0.25000
wtk2 0.09375 0.09375 0.09375 0.18750 0.09375 0.09375
0.09375 0.18750 0.03125 0.03125
wtk3 0.02778 0.02778 0.02778 0.05556 0.02778 0.02778
0.05556 0.02778 0.05556 0.05556 0.02778 0.02778
0.02778 0.05556 0.02778 0.05556 0.05556 0.05556
0.02778 0.02778 0.02778 0.02778 0.02778 0.05556
0.05556 0.00926 0.00926 0.00926
wtk4 0.01172 0.01172 0.01172 0.02344 0.01172 0.01172
0.02344 0.01172 0.02344 0.02344 0.01172 0.01172
0.02344 0.01172 0.02344 0.02344 0.01172 0.02344
0.02344 0.02344 0.01172 0.01172 0.01172 0.02344
0.01172 0.02344 0.02344 0.02344 0.01172 0.02344
0.02344 0.02344 0.02344 0.01172 0.01172 0.01172
0.01172 0.01172 0.02344 0.02344 0.01172 0.02344
0.02344 0.02344 0.02344 0.02344 0.01172 0.01172
0.01172 0.01172
outvars : Printing only first 50 k-points.
znucl 13.00000
================================================================================
chkinp: Checking input parameters for consistency, jdtset= 1.
chkinp: Checking input parameters for consistency, jdtset= 2.
chkinp: Checking input parameters for consistency, jdtset= 3.
chkinp: Checking input parameters for consistency, jdtset= 4.
================================================================================
== DATASET 1 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 1, }
dimensions: {natom: 1, nkpt: 2, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 90, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 5.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--- Pseudopotential description ------------------------------------------------
- pspini: atom type 1 psp file is /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/Pspdir/Psdj_nc_sr_04_pw_std_psp8/Al.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/Pspdir/Psdj_nc_sr_04_pw_std_psp8/Al.psp8
- Al ONCVPSP-3.3.0 r_core= 1.76802 1.76802 1.70587
- 13.00000 3.00000 171102 znucl, zion, pspdat
8 -1012 2 4 600 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
5.99000000000000 5.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2 2
extension_switch 1
pspatm : epsatm= 0.57439192
--- l ekb(1:nproj) -->
0 5.725870 0.726131
1 6.190420 0.914022
2 -4.229503 -0.925599
pspatm: atomic psp has been read and splines computed
1.72317576E+00 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
_setup2: Arith. and geom. avg. npw (full set) are 89.750 89.749
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3578817289599 -2.358E+00 1.732E-03 2.558E-01
ETOT 2 -2.3580384196975 -1.567E-04 2.687E-08 1.111E-02
ETOT 3 -2.3580435144617 -5.095E-06 1.068E-07 3.747E-05
ETOT 4 -2.3580435382090 -2.375E-08 7.790E-10 1.373E-07
ETOT 5 -2.3580435382703 -6.130E-11 1.413E-12 1.561E-09
ETOT 6 -2.3580435382711 -8.140E-13 3.028E-14 4.181E-12
ETOT 7 -2.3580435382711 5.773E-15 1.204E-16 3.653E-14
ETOT 8 -2.3580435382711 6.217E-15 1.855E-18 5.322E-17
At SCF step 8 vres2 = 5.32E-17 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.58898574E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.58898574E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.58898574E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 1, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: 6.217E-15, res2: 5.322E-17, residm: 1.855E-18, diffor: null, }
etotal : -2.35804354E+00
entropy : 0.00000000E+00
fermie : 2.71850060E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -2.58898574E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -2.58898574E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -2.58898574E-06, ]
pressure_GPa: 7.6171E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93506344
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-2.58898574492111E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -2.58898574492111E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -2.58898574492111E-06
Total energy (etotal) [Ha]= -2.35804353827112E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3580435962250 -2.358E+00 8.995E-14 6.743E-08
ETOT 2 -2.3580435962413 -1.636E-11 1.134E-16 2.950E-09
ETOT 3 -2.3580435962425 -1.176E-12 2.232E-14 1.035E-11
ETOT 4 -2.3580435962425 -3.464E-14 1.561E-16 2.315E-14
ETOT 5 -2.3580435962425 -1.332E-15 2.792E-19 2.057E-16
At SCF step 5 vres2 = 2.06E-16 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.94411356E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.94411356E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.94411356E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 1, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8002951, 3.8002951, ]
- [ 3.8002951, 0.0000000, 3.8002951, ]
- [ 3.8002951, 3.8002951, 0.0000000, ]
lattice_lengths: [ 5.37443, 5.37443, 5.37443, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0976957E+02
convergence: {deltae: -1.332E-15, res2: 2.057E-16, residm: 2.792E-19, diffor: null, }
etotal : -2.35804360E+00
entropy : 0.00000000E+00
fermie : 2.71766783E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.94411356E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.94411356E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.94411356E-06, ]
pressure_GPa: 5.7198E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93518138
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60059028874984E+00 7.60059028874984E+00 7.60059028874984E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80029514437492E+00 3.80029514437492E+00
3.80029514437492E+00 0.00000000000000E+00 3.80029514437492E+00
3.80029514437492E+00 3.80029514437492E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09769573294807E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37442893419563E+00 5.37442893419563E+00 5.37442893419563E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.94411355938651E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.94411355938654E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.94411355938654E-06
Total energy (etotal) [Ha]= -2.35804359624255E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-5.79714E-08
Relative =-2.45845E-08
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3580436714834 -2.358E+00 8.043E-13 6.143E-07
ETOT 2 -2.3580436716335 -1.501E-10 3.173E-15 2.686E-08
ETOT 3 -2.3580436716444 -1.084E-11 2.034E-13 9.444E-11
ETOT 4 -2.3580436716444 -5.240E-14 2.218E-15 2.112E-13
ETOT 5 -2.3580436716445 -1.288E-14 2.481E-18 1.861E-15
At SCF step 5 vres2 = 1.86E-15 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.20376241E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.20376241E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.20376241E-08 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 1, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8011858, 3.8011858, ]
- [ 3.8011858, 0.0000000, 3.8011858, ]
- [ 3.8011858, 3.8011858, 0.0000000, ]
lattice_lengths: [ 5.37569, 5.37569, 5.37569, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0984677E+02
convergence: {deltae: -1.288E-14, res2: 1.861E-15, residm: 2.481E-18, diffor: null, }
etotal : -2.35804367E+00
entropy : 0.00000000E+00
fermie : 2.71515626E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.20376241E-08, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.20376241E-08, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.20376241E-08, ]
pressure_GPa: 3.5416E-04
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93553740
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60237151417214E+00 7.60237151417214E+00 7.60237151417214E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80118575708607E+00 3.80118575708607E+00
3.80118575708607E+00 0.00000000000000E+00 3.80118575708607E+00
3.80118575708607E+00 3.80118575708607E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09846766054524E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37568845077056E+00 5.37568845077056E+00 5.37568845077056E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.20376241298850E-08 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.20376241298308E-08 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.20376241298579E-08
Total energy (etotal) [Ha]= -2.35804367164445E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-7.54019E-08
Relative =-3.19765E-08
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 1.2038E-06 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 16.085E-19; max= 24.806E-19
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.602371514172 7.602371514172 7.602371514172 bohr
= 4.023001751389 4.023001751389 4.023001751389 angstroms
prteigrs : about to open file tbase4_2o_DS1_EIG
Fermi (or HOMO) energy (hartree) = 0.27152 Average Vxc (hartree)= -0.36713
Eigenvalues (hartree) for nkpt= 2 k points:
kpt# 1, nband= 3, wtk= 0.75000, kpt= -0.2500 0.5000 0.0000 (reduced coord)
0.09836 0.25743 0.42131
occupation numbers for kpt# 1
2.00003 1.33305 0.00015
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 1, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.68011933496529E-01
hartree : 3.75146400382928E-03
xc : -1.11506250787288E+00
Ewald energy : -2.71387412402180E+00
psp_core : 1.56870868639818E-02
local_psp : 1.66224848969190E-01
non_local_psp : 4.25216125769966E-01
internal : -2.35004517279118E+00
'-kT*entropy' : -7.99849885327223E-03
total_energy : -2.35804367164445E+00
total_energy_eV : -6.41656371340084E+01
band_energy : 3.72515000939614E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.20376241E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.20376241E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.20376241E-08 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 3.5416E-04 GPa]
- sigma(1 1)= -3.54159129E-04 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -3.54159129E-04 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -3.54159129E-04 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 2 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 2, }
dimensions: {natom: 1, nkpt: 10, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 92, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 5.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 1.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_2o_DS1_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.000 88.972
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581245516017 -2.358E+00 5.698E-01 1.977E-04
ETOT 2 -2.3581245978625 -4.626E-08 1.147E-07 8.287E-06
ETOT 3 -2.3581246009965 -3.134E-09 4.380E-09 4.153E-08
ETOT 4 -2.3581246010221 -2.559E-11 8.871E-11 9.320E-11
ETOT 5 -2.3581246010222 -5.995E-14 7.776E-12 7.600E-13
ETOT 6 -2.3581246010222 -2.665E-15 1.859E-13 3.122E-15
At SCF step 6 vres2 = 3.12E-15 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 3.84252342E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 3.84252342E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.84252342E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -2.665E-15, res2: 3.122E-15, residm: 1.859E-13, diffor: null, }
etotal : -2.35812460E+00
entropy : 0.00000000E+00
fermie : 2.88015521E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 3.84252342E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 3.84252342E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.84252342E-05, ]
pressure_GPa: -1.1305E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93069843
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
3.84252341976002E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.84252341976001E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 3.84252341976001E-05
Total energy (etotal) [Ha]= -2.35812460102219E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581374743378 -2.358E+00 5.348E-10 1.634E-05
ETOT 2 -2.3581374784184 -4.081E-09 2.101E-13 7.079E-07
ETOT 3 -2.3581374787089 -2.905E-10 1.331E-11 2.512E-09
ETOT 4 -2.3581374787104 -1.529E-12 2.033E-13 6.308E-12
ETOT 5 -2.3581374787104 -6.217E-15 5.032E-15 5.160E-14
ETOT 6 -2.3581374787104 3.997E-15 1.697E-16 1.243E-16
At SCF step 6 vres2 = 1.24E-16 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.95019565E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.95019565E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.95019565E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7956195, 3.7956195, ]
- [ 3.7956195, 0.0000000, 3.7956195, ]
- [ 3.7956195, 3.7956195, 0.0000000, ]
lattice_lengths: [ 5.36782, 5.36782, 5.36782, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0936491E+02
convergence: {deltae: 3.997E-15, res2: 1.243E-16, residm: 1.697E-16, diffor: null, }
etotal : -2.35813748E+00
entropy : 0.00000000E+00
fermie : 2.89312189E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 2.95019565E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 2.95019565E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 2.95019565E-05, ]
pressure_GPa: -8.6798E-01
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92890344
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.59123904660295E+00 7.59123904660295E+00 7.59123904660295E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.79561952330147E+00 3.79561952330147E+00
3.79561952330147E+00 0.00000000000000E+00 3.79561952330147E+00
3.79561952330147E+00 3.79561952330147E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09364912830265E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.36781660746105E+00 5.36781660746105E+00 5.36781660746105E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
2.95019564677918E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 2.95019564677917E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 2.95019564677917E-05
Total energy (etotal) [Ha]= -2.35813747871043E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.28777E-05
Relative =-5.46097E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581560346588 -2.358E+00 5.366E-09 1.715E-04
ETOT 2 -2.3581560766026 -4.194E-08 1.655E-12 7.380E-06
ETOT 3 -2.3581560795691 -2.966E-09 1.344E-10 2.604E-08
ETOT 4 -2.3581560795847 -1.568E-11 2.118E-12 6.472E-11
ETOT 5 -2.3581560795848 -4.974E-14 5.694E-14 5.210E-13
ETOT 6 -2.3581560795848 8.882E-16 2.310E-15 1.128E-15
At SCF step 6 vres2 = 1.13E-15 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.49327083E-07 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.49327083E-07 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.49327083E-07 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7813499, 3.7813499, ]
- [ 3.7813499, 0.0000000, 3.7813499, ]
- [ 3.7813499, 3.7813499, 0.0000000, ]
lattice_lengths: [ 5.34764, 5.34764, 5.34764, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0813607E+02
convergence: {deltae: 8.882E-16, res2: 1.128E-15, residm: 2.310E-15, diffor: null, }
etotal : -2.35815608E+00
entropy : 0.00000000E+00
fermie : 2.93567359E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 2.49327083E-07, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 2.49327083E-07, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 2.49327083E-07, ]
pressure_GPa: -7.3355E-03
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92309825
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.56269975058648E+00 7.56269975058648E+00 7.56269975058648E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.78134987529324E+00 3.78134987529324E+00
3.78134987529324E+00 0.00000000000000E+00 3.78134987529324E+00
3.78134987529324E+00 3.78134987529324E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.08136070680423E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.34763627771751E+00 5.34763627771751E+00 5.34763627771751E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
2.49327082551924E-07 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 2.49327082551897E-07 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 2.49327082551951E-07
Total energy (etotal) [Ha]= -2.35815607958479E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.86009E-05
Relative =-7.88792E-06
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 2.4933E-05 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 89.319E-18; max= 23.096E-16
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.562699750586 7.562699750586 7.562699750586 bohr
= 4.002008358197 4.002008358197 4.002008358197 angstroms
prteigrs : about to open file tbase4_2o_DS2_EIG
Fermi (or HOMO) energy (hartree) = 0.29357 Average Vxc (hartree)= -0.36907
Eigenvalues (hartree) for nkpt= 10 k points:
kpt# 1, nband= 3, wtk= 0.09375, kpt= -0.1250 -0.2500 0.0000 (reduced coord)
-0.06763 0.49087 0.62308
occupation numbers for kpt# 1
2.00000 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 2, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.69481856686118E-01
hartree : 4.02724204003121E-03
xc : -1.11910429756127E+00
Ewald energy : -2.72811033280968E+00
psp_core : 1.59352540737065E-02
local_psp : 1.77162662849965E-01
non_local_psp : 4.24111513874819E-01
internal : -2.35649610084631E+00
'-kT*entropy' : -1.65997873848375E-03
total_energy : -2.35815607958479E+00
total_energy_eV : -6.41686959098901E+01
band_energy : 3.79131573777803E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.49327083E-07 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.49327083E-07 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.49327083E-07 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= -7.3355E-03 GPa]
- sigma(1 1)= 7.33545602E-03 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= 7.33545602E-03 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= 7.33545602E-03 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 3 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 3, }
dimensions: {natom: 1, nkpt: 28, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 94, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 5.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 2.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_2o_DS2_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.361 89.328
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 3, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3583794421966 -2.358E+00 1.615E-03 5.012E-04
ETOT 2 -2.3583796963760 -2.542E-07 4.412E-07 2.542E-05
ETOT 3 -2.3583797118611 -1.549E-08 6.716E-08 2.885E-08
ETOT 4 -2.3583797118764 -1.527E-11 2.035E-10 2.080E-10
ETOT 5 -2.3583797118765 -9.237E-14 2.780E-11 8.759E-13
ETOT 6 -2.3583797118765 -6.661E-15 3.467E-13 1.724E-15
At SCF step 6 vres2 = 1.72E-15 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 4.84842569E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 4.84842569E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.84842569E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 3, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -6.661E-15, res2: 1.724E-15, residm: 3.467E-13, diffor: null, }
etotal : -2.35837971E+00
entropy : 0.00000000E+00
fermie : 2.86227819E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 4.84842569E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 4.84842569E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 4.84842569E-05, ]
pressure_GPa: -1.4265E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93043653
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
4.84842568961671E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 4.84842568961672E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 4.84842568961671E-05
Total energy (etotal) [Ha]= -2.35837971187648E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 3, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3584001849722 -2.358E+00 1.618E-08 2.681E-05
ETOT 2 -2.3584001915753 -6.603E-09 2.993E-11 1.170E-06
ETOT 3 -2.3584001920545 -4.792E-10 4.970E-11 4.105E-09
ETOT 4 -2.3584001920570 -2.528E-12 1.635E-12 9.902E-12
ETOT 5 -2.3584001920570 7.105E-15 8.313E-14 7.977E-14
ETOT 6 -2.3584001920570 4.441E-15 2.419E-15 1.974E-16
At SCF step 6 vres2 = 1.97E-16 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 3.70969471E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 3.70969471E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.70969471E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 3, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7944728, 3.7944728, ]
- [ 3.7944728, 0.0000000, 3.7944728, ]
- [ 3.7944728, 3.7944728, 0.0000000, ]
lattice_lengths: [ 5.36619, 5.36619, 5.36619, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0926582E+02
convergence: {deltae: 4.441E-15, res2: 1.974E-16, residm: 2.419E-15, diffor: null, }
etotal : -2.35840019E+00
entropy : 0.00000000E+00
fermie : 2.87870685E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 3.70969471E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 3.70969471E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.70969471E-05, ]
pressure_GPa: -1.0914E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92814691
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.58894558942767E+00 7.58894558942767E+00 7.58894558942767E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.79447279471384E+00 3.79447279471384E+00
3.79447279471384E+00 0.00000000000000E+00 3.79447279471384E+00
3.79447279471384E+00 3.79447279471384E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09265819136255E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.36619488834005E+00 5.36619488834005E+00 5.36619488834005E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
3.70969471205302E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.70969471205302E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 3.70969471205301E-05
Total energy (etotal) [Ha]= -2.35840019205701E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.04802E-05
Relative =-8.68397E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 3, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3584280717056 -2.358E+00 1.276E-07 2.713E-04
ETOT 2 -2.3584281367470 -6.504E-08 4.085E-10 1.174E-05
ETOT 3 -2.3584281414349 -4.688E-09 4.034E-10 4.096E-08
ETOT 4 -2.3584281414598 -2.491E-11 1.512E-11 9.781E-11
ETOT 5 -2.3584281414598 -4.441E-14 4.428E-13 7.630E-13
ETOT 6 -2.3584281414599 -1.288E-14 2.026E-14 1.706E-15
At SCF step 6 vres2 = 1.71E-15 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -8.04239818E-07 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -8.04239818E-07 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -8.04239818E-07 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 3, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7767959, 3.7767959, ]
- [ 3.7767959, 0.0000000, 3.7767959, ]
- [ 3.7767959, 3.7767959, 0.0000000, ]
lattice_lengths: [ 5.34120, 5.34120, 5.34120, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0774585E+02
convergence: {deltae: -1.288E-14, res2: 1.706E-15, residm: 2.026E-14, diffor: null, }
etotal : -2.35842814E+00
entropy : 0.00000000E+00
fermie : 2.93173063E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -8.04239818E-07, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -8.04239818E-07, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -8.04239818E-07, ]
pressure_GPa: 2.3662E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92088259
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.55359189539082E+00 7.55359189539082E+00 7.55359189539082E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77679594769541E+00 3.77679594769541E+00
3.77679594769541E+00 0.00000000000000E+00 3.77679594769541E+00
3.77679594769541E+00 3.77679594769541E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07745852080294E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.34119605154659E+00 5.34119605154659E+00 5.34119605154659E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-8.04239817727554E-07 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -8.04239817727608E-07 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -8.04239817727635E-07
Total energy (etotal) [Ha]= -2.35842814145986E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.79494E-05
Relative =-1.18509E-05
--- Iteration: ( 4/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 3, itime: 4, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3584281537084 -2.358E+00 3.187E-10 1.235E-07
ETOT 2 -2.3584281537387 -3.023E-11 1.065E-13 5.380E-09
ETOT 3 -2.3584281537408 -2.154E-12 1.889E-13 1.803E-11
ETOT 4 -2.3584281537408 -2.798E-14 6.929E-15 4.394E-14
ETOT 5 -2.3584281537409 -1.021E-14 1.013E-16 3.446E-16
At SCF step 5 vres2 = 3.45E-16 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.88517645E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.88517645E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.88517645E-08 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 3, itime: 4, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7771659, 3.7771659, ]
- [ 3.7771659, 0.0000000, 3.7771659, ]
- [ 3.7771659, 3.7771659, 0.0000000, ]
lattice_lengths: [ 5.34172, 5.34172, 5.34172, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0777752E+02
convergence: {deltae: -1.021E-14, res2: 3.446E-16, residm: 1.013E-16, diffor: null, }
etotal : -2.35842815E+00
entropy : 0.00000000E+00
fermie : 2.93061320E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 2.88517645E-08, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 2.88517645E-08, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 2.88517645E-08, ]
pressure_GPa: -8.4885E-04
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92103372
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.55433186180140E+00 7.55433186180140E+00 7.55433186180140E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77716593090070E+00 3.77716593090070E+00
3.77716593090070E+00 0.00000000000000E+00 3.77716593090070E+00
3.77716593090070E+00 3.77716593090070E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07777520241536E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.34171928681337E+00 5.34171928681337E+00 5.34171928681337E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
2.88517645157785E-08 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 2.88517645157243E-08 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 2.88517645157243E-08
Total energy (etotal) [Ha]= -2.35842815374086E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.22810E-08
Relative =-5.20728E-09
At Broyd/MD step 4, gradients are converged :
max grad (force/stress) = 2.8852E-06 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 54.152E-19; max= 10.129E-17
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.554331861801 7.554331861801 7.554331861801 bohr
= 3.997580262152 3.997580262152 3.997580262152 angstroms
prteigrs : about to open file tbase4_2o_DS3_EIG
Fermi (or HOMO) energy (hartree) = 0.29306 Average Vxc (hartree)= -0.36950
Eigenvalues (hartree) for nkpt= 28 k points:
kpt# 1, nband= 3, wtk= 0.02778, kpt= -0.0833 -0.1667 0.0000 (reduced coord)
-0.09934 0.58875 0.67024
occupation numbers for kpt# 1
2.00000 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 3, itime: 4, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.69651997036308E-01
hartree : 3.94653980211436E-03
xc : -1.11990242808467E+00
Ewald energy : -2.73113224451228E+00
psp_core : 1.59882669128319E-02
local_psp : 1.79201785201710E-01
non_local_psp : 4.24578924409453E-01
internal : -2.35766715923453E+00
'-kT*entropy' : -7.60994506328480E-04
total_energy : -2.35842815374086E+00
total_energy_eV : -6.41760994248384E+01
band_energy : 3.80649708738326E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.88517645E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.88517645E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.88517645E-08 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= -8.4885E-04 GPa]
- sigma(1 1)= 8.48848218E-04 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= 8.48848218E-04 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= 8.48848218E-04 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 4 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 4, }
dimensions: {natom: 1, nkpt: 60, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 95, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 5.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 3.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_2o_DS3_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.363 89.331
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 4, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3585801316737 -2.359E+00 1.017E+00 9.298E-04
ETOT 2 -2.3585804466992 -3.150E-07 2.397E-05 4.511E-05
ETOT 3 -2.3585804686186 -2.192E-08 8.968E-08 1.115E-07
ETOT 4 -2.3585804686848 -6.622E-11 2.115E-08 3.763E-10
ETOT 5 -2.3585804686850 -2.260E-13 1.571E-10 2.885E-12
ETOT 6 -2.3585804686850 -4.441E-15 2.090E-11 5.407E-15
At SCF step 6 vres2 = 5.41E-15 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 4.90828343E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 4.90828343E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.90828343E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 4, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -4.441E-15, res2: 5.407E-15, residm: 2.090E-11, diffor: null, }
etotal : -2.35858047E+00
entropy : 0.00000000E+00
fermie : 2.85831852E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 4.90828343E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 4.90828343E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 4.90828343E-05, ]
pressure_GPa: -1.4441E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93052926
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
4.90828342587702E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 4.90828342587702E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 4.90828342587701E-05
Total energy (etotal) [Ha]= -2.35858046868504E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 4, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3586014917105 -2.359E+00 5.822E-08 2.764E-05
ETOT 2 -2.3586014985045 -6.794E-09 6.004E-10 1.207E-06
ETOT 3 -2.3586014989994 -4.949E-10 6.398E-11 4.236E-09
ETOT 4 -2.3586014990020 -2.648E-12 2.659E-12 1.021E-11
ETOT 5 -2.3586014990020 8.882E-16 6.788E-14 8.157E-14
ETOT 6 -2.3586014990020 -4.441E-15 6.764E-15 2.029E-16
At SCF step 6 vres2 = 2.03E-16 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 3.77859941E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 3.77859941E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.77859941E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 4, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7944046, 3.7944046, ]
- [ 3.7944046, 0.0000000, 3.7944046, ]
- [ 3.7944046, 3.7944046, 0.0000000, ]
lattice_lengths: [ 5.36610, 5.36610, 5.36610, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0925992E+02
convergence: {deltae: -4.441E-15, res2: 2.029E-16, residm: 6.764E-15, diffor: null, }
etotal : -2.35860150E+00
entropy : 0.00000000E+00
fermie : 2.87497853E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 3.77859941E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 3.77859941E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.77859941E-05, ]
pressure_GPa: -1.1117E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92820693
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.58880911378900E+00 7.58880911378900E+00 7.58880911378900E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.79440455689450E+00 3.79440455689450E+00
3.79440455689450E+00 0.00000000000000E+00 3.79440455689450E+00
3.79440455689450E+00 3.79440455689450E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09259924303789E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.36609838549048E+00 5.36609838549048E+00 5.36609838549048E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
3.77859941050962E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.77859941050962E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 3.77859941050962E-05
Total energy (etotal) [Ha]= -2.35860149900204E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.10303E-05
Relative =-8.91647E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 4, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3586310115624 -2.359E+00 3.256E-05 2.942E-04
ETOT 2 -2.3586310818604 -7.030E-08 4.113E-09 1.274E-05
ETOT 3 -2.3586310869453 -5.085E-09 6.733E-10 4.442E-08
ETOT 4 -2.3586310869724 -2.709E-11 2.555E-11 1.057E-10
ETOT 5 -2.3586310869725 -8.793E-14 6.516E-13 8.178E-13
ETOT 6 -2.3586310869725 -3.553E-15 5.655E-14 1.834E-15
At SCF step 6 vres2 = 1.83E-15 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -9.86281103E-07 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -9.86281103E-07 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -9.86281103E-07 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 4, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7760422, 3.7760422, ]
- [ 3.7760422, 0.0000000, 3.7760422, ]
- [ 3.7760422, 3.7760422, 0.0000000, ]
lattice_lengths: [ 5.34013, 5.34013, 5.34013, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0768136E+02
convergence: {deltae: -3.553E-15, res2: 1.834E-15, residm: 5.655E-14, diffor: null, }
etotal : -2.35863109E+00
entropy : 0.00000000E+00
fermie : 2.93014056E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -9.86281103E-07, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -9.86281103E-07, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -9.86281103E-07, ]
pressure_GPa: 2.9017E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92064935
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.55208445780947E+00 7.55208445780947E+00 7.55208445780947E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77604222890473E+00 3.77604222890473E+00
3.77604222890473E+00 0.00000000000000E+00 3.77604222890473E+00
3.77604222890473E+00 3.77604222890473E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07681357835338E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.34013013221061E+00 5.34013013221061E+00 5.34013013221061E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-9.86281102539758E-07 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -9.86281102539622E-07 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -9.86281102539704E-07
Total energy (etotal) [Ha]= -2.35863108697250E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.95880E-05
Relative =-1.25446E-05
--- Iteration: ( 4/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 4, itime: 4, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {tolvrs: 1.00E-14, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3586311058377 -2.359E+00 6.347E-10 1.927E-07
ETOT 2 -2.3586311058848 -4.702E-11 5.075E-12 8.400E-09
ETOT 3 -2.3586311058882 -3.396E-12 4.536E-13 2.808E-11
ETOT 4 -2.3586311058882 -1.776E-15 1.650E-14 6.829E-14
ETOT 5 -2.3586311058882 -1.732E-14 3.708E-16 5.316E-16
At SCF step 5 vres2 = 5.32E-16 < tolvrs= 1.00E-14 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.65242360E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.65242360E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.65242360E-08 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 4, itime: 4, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7765027, 3.7765027, ]
- [ 3.7765027, 0.0000000, 3.7765027, ]
- [ 3.7765027, 3.7765027, 0.0000000, ]
lattice_lengths: [ 5.34078, 5.34078, 5.34078, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0772076E+02
convergence: {deltae: -1.732E-14, res2: 5.316E-16, residm: 3.708E-16, diffor: null, }
etotal : -2.35863111E+00
entropy : 0.00000000E+00
fermie : 2.92874782E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 2.65242360E-08, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 2.65242360E-08, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 2.65242360E-08, ]
pressure_GPa: -7.8037E-04
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92083772
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.55300549343894E+00 7.55300549343894E+00 7.55300549343894E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77650274671947E+00 3.77650274671947E+00
3.77650274671947E+00 0.00000000000000E+00 3.77650274671947E+00
3.77650274671947E+00 3.77650274671947E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07720760385912E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.34078140274992E+00 5.34078140274992E+00 5.34078140274992E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
2.65242360339261E-08 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 2.65242360338990E-08 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 2.65242360339261E-08
Total energy (etotal) [Ha]= -2.35863110588818E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.89157E-08
Relative =-8.01977E-09
At Broyd/MD step 4, gradients are converged :
max grad (force/stress) = 2.6524E-06 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 13.170E-18; max= 37.075E-17
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.553005493439 7.553005493439 7.553005493439 bohr
= 3.996878378242 3.996878378242 3.996878378242 angstroms
prteigrs : about to open file tbase4_2o_DS4_EIG
Fermi (or HOMO) energy (hartree) = 0.29287 Average Vxc (hartree)= -0.36957
Eigenvalues (hartree) for nkpt= 60 k points:
kpt# 1, nband= 3, wtk= 0.01172, kpt= -0.0625 -0.1250 0.0000 (reduced coord)
-0.11067 0.64002 0.69698
occupation numbers for kpt# 1
2.00000 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 4, itime: 4, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.69270921934112E-01
hartree : 3.92641908818661E-03
xc : -1.12003266025629E+00
Ewald energy : -2.73161185324630E+00
psp_core : 1.59966913958971E-02
local_psp : 1.79433341494374E-01
non_local_psp : 4.24814346194203E-01
internal : -2.35820279339583E+00
'-kT*entropy' : -4.28312492349626E-04
total_energy : -2.35863110588818E+00
total_energy_eV : -6.41816220341085E+01
band_energy : 3.80538664090510E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.65242360E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.65242360E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.65242360E-08 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= -7.8037E-04 GPa]
- sigma(1 1)= 7.80369966E-04 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= 7.80369966E-04 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= 7.80369966E-04 sigma(2 1)= 0.00000000E+00
== END DATASET(S) ==============================================================
================================================================================
-outvars: echo values of variables after computation --------
acell1 7.6023715142E+00 7.6023715142E+00 7.6023715142E+00 Bohr
acell2 7.5626997506E+00 7.5626997506E+00 7.5626997506E+00 Bohr
acell3 7.5543318618E+00 7.5543318618E+00 7.5543318618E+00 Bohr
acell4 7.5530054934E+00 7.5530054934E+00 7.5530054934E+00 Bohr
amu 2.69815390E+01
dilatmx 1.05000000E+00
ecut 6.00000000E+00 Hartree
ecutsm 5.00000000E-01 Hartree
etotal1 -2.3580436716E+00
etotal2 -2.3581560796E+00
etotal3 -2.3584281537E+00
etotal4 -2.3586311059E+00
fcart1 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart2 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart3 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart4 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
- fftalg 512
getwfk -1
ionmov 2
ixc -1012
jdtset 1 2 3 4
kpt1 -2.50000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
kpt2 -1.25000000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.75000000E-01 0.00000000E+00
-3.75000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 1.25000000E-01
-1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
kpt3 -8.33333333E-02 -1.66666667E-01 0.00000000E+00
-8.33333333E-02 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -2.50000000E-01 0.00000000E+00
-8.33333333E-02 -2.50000000E-01 8.33333333E-02
-8.33333333E-02 5.00000000E-01 0.00000000E+00
-1.66666667E-01 -4.16666667E-01 0.00000000E+00
-8.33333333E-02 -4.16666667E-01 8.33333333E-02
-2.50000000E-01 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -3.33333333E-01 8.33333333E-02
-8.33333333E-02 -3.33333333E-01 1.66666667E-01
-8.33333333E-02 3.33333333E-01 0.00000000E+00
-1.66666667E-01 4.16666667E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 0.00000000E+00
-1.66666667E-01 5.00000000E-01 8.33333333E-02
-3.33333333E-01 -4.16666667E-01 0.00000000E+00
-2.50000000E-01 -4.16666667E-01 8.33333333E-02
-1.66666667E-01 -4.16666667E-01 1.66666667E-01
-8.33333333E-02 -4.16666667E-01 2.50000000E-01
-8.33333333E-02 1.66666667E-01 0.00000000E+00
-1.66666667E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.33333333E-01 0.00000000E+00
-3.33333333E-01 4.16666667E-01 0.00000000E+00
-4.16666667E-01 5.00000000E-01 0.00000000E+00
-3.33333333E-01 5.00000000E-01 8.33333333E-02
-2.50000000E-01 5.00000000E-01 1.66666667E-01
-8.33333333E-02 0.00000000E+00 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
-4.16666667E-01 0.00000000E+00 0.00000000E+00
kpt4 -6.25000000E-02 -1.25000000E-01 0.00000000E+00
-6.25000000E-02 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 -1.87500000E-01 0.00000000E+00
-6.25000000E-02 -1.87500000E-01 6.25000000E-02
-6.25000000E-02 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.12500000E-01 0.00000000E+00
-6.25000000E-02 -3.12500000E-01 6.25000000E-02
-1.87500000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 -2.50000000E-01 6.25000000E-02
-6.25000000E-02 -2.50000000E-01 1.25000000E-01
-6.25000000E-02 5.00000000E-01 0.00000000E+00
-1.25000000E-01 -4.37500000E-01 0.00000000E+00
-6.25000000E-02 -4.37500000E-01 6.25000000E-02
-1.87500000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 6.25000000E-02
-6.25000000E-02 -3.75000000E-01 1.25000000E-01
-2.50000000E-01 -3.12500000E-01 0.00000000E+00
-1.87500000E-01 -3.12500000E-01 6.25000000E-02
-1.25000000E-01 -3.12500000E-01 1.25000000E-01
-6.25000000E-02 -3.12500000E-01 1.87500000E-01
-6.25000000E-02 3.75000000E-01 0.00000000E+00
-1.25000000E-01 4.37500000E-01 0.00000000E+00
-1.87500000E-01 5.00000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 6.25000000E-02
-2.50000000E-01 -4.37500000E-01 0.00000000E+00
-1.87500000E-01 -4.37500000E-01 6.25000000E-02
-1.25000000E-01 -4.37500000E-01 1.25000000E-01
-6.25000000E-02 -4.37500000E-01 1.87500000E-01
-3.12500000E-01 -3.75000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 6.25000000E-02
-1.87500000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 -3.75000000E-01 1.87500000E-01
-6.25000000E-02 -3.75000000E-01 2.50000000E-01
-6.25000000E-02 2.50000000E-01 0.00000000E+00
-1.25000000E-01 3.12500000E-01 0.00000000E+00
-1.87500000E-01 3.75000000E-01 0.00000000E+00
-2.50000000E-01 4.37500000E-01 0.00000000E+00
-3.12500000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 6.25000000E-02
-1.87500000E-01 5.00000000E-01 1.25000000E-01
-3.75000000E-01 -4.37500000E-01 0.00000000E+00
-3.12500000E-01 -4.37500000E-01 6.25000000E-02
-2.50000000E-01 -4.37500000E-01 1.25000000E-01
-1.87500000E-01 -4.37500000E-01 1.87500000E-01
-1.25000000E-01 -4.37500000E-01 2.50000000E-01
-6.25000000E-02 -4.37500000E-01 3.12500000E-01
-6.25000000E-02 1.25000000E-01 0.00000000E+00
-1.25000000E-01 1.87500000E-01 0.00000000E+00
-1.87500000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.12500000E-01 0.00000000E+00
outvar_i_n : Printing only first 50 k-points.
kptrlatt1 2 -2 2 -2 2 2 -2 -2 2
kptrlatt2 4 -4 4 -4 4 4 -4 -4 4
kptrlatt3 6 -6 6 -6 6 6 -6 -6 6
kptrlatt4 8 -8 8 -8 8 8 -8 -8 8
kptrlen1 1.52000000E+01
kptrlen2 3.04000000E+01
kptrlen3 4.56000000E+01
kptrlen4 6.08000000E+01
P mkmem1 2
P mkmem2 10
P mkmem3 28
P mkmem4 60
natom 1
nband1 3
nband2 3
nband3 3
nband4 3
ndtset 4
ngfft 15 15 15
nkpt1 2
nkpt2 10
nkpt3 28
nkpt4 60
nstep 10
nsym 48
ntime 10
ntypat 1
occ1 2.000034 1.333048 0.000150
2.000000 0.000306 0.000000
occ2 2.000000 0.000000 0.000000
2.000001 2.013713 0.000000
2.000000 0.001820 0.000000
2.000000 0.741556 0.000017
2.000000 0.004812 0.000000
2.000282 2.110043 0.005108
2.000242 0.471370 0.004673
2.011314 1.952546 0.004209
2.000000 0.000000 0.000000
2.000000 1.905956 0.000000
occ3 2.000000 0.000000 0.000000
2.000000 0.000100 0.000000
2.000000 0.000000 0.000000
2.000000 0.000280 0.000000
2.000004 2.002472 0.000000
2.000000 0.619602 0.000000
2.000000 2.140780 0.000000
2.000000 0.000009 0.000000
2.000000 0.005229 0.000000
2.000000 0.269397 0.000348
2.000000 1.200974 0.000000
2.000041 2.011308 0.000000
2.000004 1.779691 0.000471
2.000341 2.038448 0.000015
2.000000 0.004244 0.000329
2.000000 0.001895 0.001812
2.000041 1.338799 0.003800
2.010299 2.136265 0.003284
2.000000 0.000000 0.000000
2.000000 0.002924 0.000001
2.000004 1.850594 0.005260
2.029838 2.085036 0.313392
2.001722 1.494477 0.001330
2.008361 0.919422 0.043313
2.077759 1.860814 0.088381
2.000000 0.000000 0.000000
2.000000 0.000893 0.000000
2.000000 2.093129 0.000000
occ4 2.000000 0.000000 0.000000
2.000000 0.000043 0.000000
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 0.974189 0.000000
2.000000 0.002535 0.000000
2.000000 0.002194 0.000000
2.000000 0.000000 0.000000
2.000000 0.000007 0.000000
2.000000 0.001880 0.000000
2.000006 2.000995 0.000000
2.000000 2.052906 0.000000
2.000000 2.052019 0.000000
2.000000 0.002902 0.000000
2.000000 0.395216 0.000000
2.000000 1.907174 0.000000
2.000000 0.000000 0.000000
2.000000 0.000467 0.000000
2.000000 0.004270 0.000009
2.000000 0.132334 0.001020
2.000000 2.127608 0.000000
2.000025 2.002677 0.000000
2.000002 2.117398 0.000000
2.000089 2.006538 0.000000
2.000000 0.026370 0.000016
2.000000 1.361309 0.000002
2.000007 2.139839 0.000006
2.000858 2.028176 0.000321
2.000000 0.000151 0.000010
2.000000 0.003322 0.000055
2.000000 0.009667 0.000276
2.000002 0.836111 0.004685
2.000301 1.992955 0.000304
2.000000 0.003850 0.000000
2.000000 0.663953 0.000000
2.000002 2.115826 0.000015
2.002221 2.052107 0.002609
2.000024 0.230562 0.002984
2.000281 1.916129 0.005132
2.005517 2.082543 0.001664
2.000001 0.004653 0.000093
2.000006 0.000016 0.005248
2.000086 0.139403 0.004633
2.002299 1.518590 0.003294
2.046821 2.140549 0.054449
2.064172 2.072589 0.547684
2.000000 0.000000 0.000000
2.000000 0.000023 0.000000
2.000000 0.001566 0.000026
2.000000 1.181120 0.005259
prtocc : prtvol=0, do not print more k-points.
occopt 4
optcell 1
rprim 0.0000000000E+00 5.0000000000E-01 5.0000000000E-01
5.0000000000E-01 0.0000000000E+00 5.0000000000E-01
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
shiftk 5.00000000E-01 5.00000000E-01 5.00000000E-01
spgroup 225
strten1 -1.2037624130E-08 -1.2037624130E-08 -1.2037624130E-08
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten2 2.4932708255E-07 2.4932708255E-07 2.4932708255E-07
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten3 2.8851764516E-08 2.8851764516E-08 2.8851764516E-08
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten4 2.6524236034E-08 2.6524236034E-08 2.6524236034E-08
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
tolvrs 1.00000000E-14
tsmear 5.00000000E-02 Hartree
typat 1
wtk1 0.75000 0.25000
wtk2 0.09375 0.09375 0.09375 0.18750 0.09375 0.09375
0.09375 0.18750 0.03125 0.03125
wtk3 0.02778 0.02778 0.02778 0.05556 0.02778 0.02778
0.05556 0.02778 0.05556 0.05556 0.02778 0.02778
0.02778 0.05556 0.02778 0.05556 0.05556 0.05556
0.02778 0.02778 0.02778 0.02778 0.02778 0.05556
0.05556 0.00926 0.00926 0.00926
wtk4 0.01172 0.01172 0.01172 0.02344 0.01172 0.01172
0.02344 0.01172 0.02344 0.02344 0.01172 0.01172
0.02344 0.01172 0.02344 0.02344 0.01172 0.02344
0.02344 0.02344 0.01172 0.01172 0.01172 0.02344
0.01172 0.02344 0.02344 0.02344 0.01172 0.02344
0.02344 0.02344 0.02344 0.01172 0.01172 0.01172
0.01172 0.01172 0.02344 0.02344 0.01172 0.02344
0.02344 0.02344 0.02344 0.02344 0.01172 0.01172
0.01172 0.01172
outvars : Printing only first 50 k-points.
znucl 13.00000
================================================================================
- Timing analysis has been suppressed with timopt=0
================================================================================
Suggested references for the acknowledgment of ABINIT usage.
The users of ABINIT have little formal obligations with respect to the ABINIT group
(those specified in the GNU General Public License, http://www.gnu.org/copyleft/gpl.txt).
However, it is common practice in the scientific literature,
to acknowledge the efforts of people that have made the research possible.
In this spirit, please find below suggested citations of work written by ABINIT developers,
corresponding to implementations inside of ABINIT that you have used in the present run.
Note also that it will be of great value to readers of publications presenting these results,
to read papers enabling them to understand the theoretical formalism and details
of the ABINIT implementation.
For information on why they are suggested, see also https://docs.abinit.org/theory/acknowledgments.
-
- [1] Libxc: A library of exchange and correlation functionals for density functional theory.
- M.A.L. Marques, M.J.T. Oliveira, T. Burnus, Computer Physics Communications 183, 2227 (2012).
- Comment: to be cited when LibXC is used (negative value of ixc)
- Strong suggestion to cite this paper.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#marques2012
-
- [2] The Abinit project: Impact, environment and recent developments.
- Computer Phys. Comm. 248, 107042 (2020).
- X.Gonze, B. Amadon, G. Antonius, F.Arnardi, L.Baguet, J.-M.Beuken,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, N.Brouwer, F.Bruneval,
- G.Brunin, T.Cavignac, J.-B. Charraud, Wei Chen, M.Cote, S.Cottenier,
- J.Denier, G.Geneste, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, Xu He, N.Helbig, N.Holzwarth, Y.Jia, F.Jollet,
- W.Lafargue-Dit-Hauret, K.Lejaeghere, M.A.L.Marques, A.Martin, C.Martins,
- H.P.C. Miranda, F.Naccarato, K. Persson, G.Petretto, V.Planes, Y.Pouillon,
- S.Prokhorenko, F.Ricci, G.-M.Rignanese, A.H.Romero, M.M.Schmitt, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, G.Zerah and J.W.Zwanzig
- Comment: the fifth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT20.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2020
-
- [3] Optimized norm-conserving Vanderbilt pseudopotentials.
- D.R. Hamann, Phys. Rev. B 88, 085117 (2013).
- Comment: Some pseudopotential generated using the ONCVPSP code were used.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#hamann2013
-
- [4] ABINIT: Overview, and focus on selected capabilities
- J. Chem. Phys. 152, 124102 (2020).
- A. Romero, D.C. Allan, B. Amadon, G. Antonius, T. Applencourt, L.Baguet,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, F.Bruneval,
- G.Brunin, D.Caliste, M.Cote,
- J.Denier, C. Dreyer, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, F.Jollet, G. Jomard,
- A.Martin,
- H.P.C. Miranda, F.Naccarato, G.Petretto, N.A. Pike, V.Planes,
- S.Prokhorenko, T. Rangel, F.Ricci, G.-M.Rignanese, M.Royo, M.Stengel, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, J.Wiktor, J.W.Zwanziger, and X.Gonze.
- Comment: a global overview of ABINIT, with focus on selected capabilities .
- Note that a version of this paper, that is not formatted for J. Chem. Phys
- is available at https://www.abinit.org/sites/default/files/ABINIT20_JPC.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#romero2020
-
- [5] Recent developments in the ABINIT software package.
- Computer Phys. Comm. 205, 106 (2016).
- X.Gonze, F.Jollet, F.Abreu Araujo, D.Adams, B.Amadon, T.Applencourt,
- C.Audouze, J.-M.Beuken, J.Bieder, A.Bokhanchuk, E.Bousquet, F.Bruneval
- D.Caliste, M.Cote, F.Dahm, F.Da Pieve, M.Delaveau, M.Di Gennaro,
- B.Dorado, C.Espejo, G.Geneste, L.Genovese, A.Gerossier, M.Giantomassi,
- Y.Gillet, D.R.Hamann, L.He, G.Jomard, J.Laflamme Janssen, S.Le Roux,
- A.Levitt, A.Lherbier, F.Liu, I.Lukacevic, A.Martin, C.Martins,
- M.J.T.Oliveira, S.Ponce, Y.Pouillon, T.Rangel, G.-M.Rignanese,
- A.H.Romero, B.Rousseau, O.Rubel, A.A.Shukri, M.Stankovski, M.Torrent,
- M.J.Van Setten, B.Van Troeye, M.J.Verstraete, D.Waroquier, J.Wiktor,
- B.Xu, A.Zhou, J.W.Zwanziger.
- Comment: the fourth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT16.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2016
-
- Proc. 0 individual time (sec): cpu= 4.0 wall= 4.1
================================================================================
Calculation completed.
.Delivered 49 WARNINGs and 32 COMMENTs to log file.
+Overall time at end (sec) : cpu= 4.0 wall= 4.1
The run might take a few seconds on a modern PC.
You will see that, for the particular value tsmear = 0.05 Ha, the lattice parameter is already converged with nkpt = 10:
acell1 7.6023827082E+00 7.6023827082E+00 7.6023827082E+00 Bohr acell2 7.5627822506E+00 7.5627822506E+00 7.5627822506E+00 Bohr acell3 7.5543007304E+00 7.5543007304E+00 7.5543007304E+00 Bohr acell4 7.5529744581E+00 7.5529744581E+00 7.5529744581E+00 Bohr
Note that there is usually a strong cross-convergence effect between the number of k-points and the value of the broadening, tsmear. The right procedure is: for each value of tsmear, convergence with respect to the number of k-points, then compare the k-point converged values for different values of tsmear.
In what follows, we will restrict ourselves to the grids with nkpt = 2, 10 and 28.
The convergence study with respect to both number of k-points and broadening factor¶
The theoretical convergence rate as a function of tsmear heading to 0, in the case of occopt = 4, is cubic. We rely on this value of occopt for this tutorial. Still, it might not be always robust, as this value might yield difficulties to find univocally the Fermi energy. A slightly worse convergence rate (quadratic) is obtained with occopt = 7, which is actually the recommended value for metallic systems.
Such convergence rates are obtained in the hypothesis of infinitely dense k-point grid. We will check the evolution of acell as a function of tsmear, for the following values of tsmear: 0.01, 0.02, 0.03 and 0.04.
Use the double-loop capability of the multi-dataset mode, with the tsmear changes in the inner loop. This will saves CPU time, as the wavefunctions of the previous dataset will be excellent (no transfer to different k-points).
The input file tbase4_3.abi is an example:
# Crystalline aluminum : computation of the total energy # # Convergence with respect to k points ndtset 12 udtset 3 4 getwfk -1 #Definition of the unit cell acell 3*7.60 # This is equivalent to 7.60 7.60 7.60 rprim 0.0 0.5 0.5 # FCC primitive vectors (to be scaled by acell) 0.5 0.0 0.5 0.5 0.5 0.0 #Definition of the atom types ntypat 1 # There is only one type of atom znucl 13 # The keyword "znucl" refers to the atomic number of the # possible type(s) of atom. The pseudopotential(s) # mentioned in the "files" file must correspond # to the type(s) of atom. Here, the only type is Aluminum pp_dirpath "$ABI_PSPDIR" # This is the path to the directory were # pseudopotentials for tests are stored pseudos "Psdj_nc_sr_04_pw_std_psp8/Al.psp8" # Name and location of the pseudopotential #Definition of the atoms natom 1 # There is only one atom per cell typat 1 # This atom is of type 1, that is, Aluminum xred # This keyword indicate that the location of the atoms # will follow, one triplet of number for each atom 0.0 0.0 0.0 # Triplet giving the REDUCED coordinate of atom 1. #Definition of the planewave basis set ecut 6.0 # Maximal kinetic energy cut-off, in Hartree #Definition of the k-point grids nshiftk 4 shiftk 0.5 0.5 0.5 # These shifts will be the same for all grids 0.5 0.0 0.0 0.0 0.5 0.0 0.0 0.0 0.5 ngkpt1? 2 2 2 ngkpt2? 4 4 4 ngkpt3? 6 6 6 #Definition of the SCF procedure nstep 10 # Maximal number of SCF cycles toldfe 1.0d-6 # Will stop when, twice in a row, the difference # between two consecutive evaluations of total energy # differ by less than toldfe (in Hartree) # This value is WAY TOO LARGE for most realistic studies of materials #Definition of occupation numbers occopt 4 tsmear?1 0.01 tsmear?2 0.02 tsmear?3 0.03 tsmear?4 0.04 #Optimization of the lattice parameters optcell 1 geoopt "bfgs" ntime 10 dilatmx 1.05 ecutsm 0.5 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% tbase4_3.abo, tolnlines = 0, tolabs = 0.0e+00, tolrel = 0.0e+00, fld_options = -easy #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = Unknown #%% keywords = #%% description = #%% Crystalline aluminum : computation of the total energy #%% #%% Convergence with respect to k points #%%<END TEST_INFO>
while tbase4_3.abo is the reference output file.
.Version 10.5.8.2 of ABINIT, released Oct 2025.
.(MPI version, prepared for a x86_64_linux_gnu13.2 computer)
.Copyright (C) 1998-2026 ABINIT group .
ABINIT comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Sat 20 Dec 2025.
- ( at 17h05 )
- input file -> /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/TestBot_MPI1/tutorial_tbase4_3/tbase4_3.abi
- output file -> tbase4_3.abo
- root for input files -> tbase4_3i
- root for output files -> tbase4_3o
DATASET 11 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 11.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 2
mpw = 90 nfft = 3375 nkpt = 2
================================================================================
P This job should need less than 2.510 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.010 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 12 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 12.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 2
mpw = 90 nfft = 3375 nkpt = 2
================================================================================
P This job should need less than 2.510 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.010 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 13 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 13.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 2
mpw = 90 nfft = 3375 nkpt = 2
================================================================================
P This job should need less than 2.510 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.010 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 14 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 14.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 2
mpw = 90 nfft = 3375 nkpt = 2
================================================================================
P This job should need less than 2.510 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.010 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 21 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 21.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 10
mpw = 92 nfft = 3375 nkpt = 10
================================================================================
P This job should need less than 2.556 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.044 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 22 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 22.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 10
mpw = 92 nfft = 3375 nkpt = 10
================================================================================
P This job should need less than 2.556 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.044 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 23 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 23.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 10
mpw = 92 nfft = 3375 nkpt = 10
================================================================================
P This job should need less than 2.556 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.044 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 24 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 24.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 10
mpw = 92 nfft = 3375 nkpt = 10
================================================================================
P This job should need less than 2.556 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.044 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 31 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 31.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 28
mpw = 94 nfft = 3375 nkpt = 28
================================================================================
P This job should need less than 2.661 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.122 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 32 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 32.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 28
mpw = 94 nfft = 3375 nkpt = 28
================================================================================
P This job should need less than 2.661 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.122 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 33 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 33.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 28
mpw = 94 nfft = 3375 nkpt = 28
================================================================================
P This job should need less than 2.661 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.122 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
DATASET 34 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 34.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 15 mpssoang = 3 mqgrid = 3001
natom = 1 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 3 mffmem = 1 mkmem = 28
mpw = 94 nfft = 3375 nkpt = 28
================================================================================
P This job should need less than 2.661 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.122 Mbytes ; DEN or POT disk file : 0.028 Mbytes.
================================================================================
--------------------------------------------------------------------------------
------------- Echo of variables that govern the present computation ------------
--------------------------------------------------------------------------------
-
- outvars: echo of selected default values
- iomode0 = 0 , fftalg0 =512 , wfoptalg0 = 0
-
- outvars: echo of global parameters not present in the input file
- max_nthreads = 0
-
-outvars: echo values of preprocessed input variables --------
acell 7.6000000000E+00 7.6000000000E+00 7.6000000000E+00 Bohr
amu 2.69815390E+01
dilatmx 1.05000000E+00
ecut 6.00000000E+00 Hartree
ecutsm 5.00000000E-01 Hartree
- fftalg 512
getwfk -1
ionmov 2
ixc -1012
jdtset 11 12 13 14 21 22 23 24 31 32
33 34
kpt11 -2.50000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
kpt12 -2.50000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
kpt13 -2.50000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
kpt14 -2.50000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
kpt21 -1.25000000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.75000000E-01 0.00000000E+00
-3.75000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 1.25000000E-01
-1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
kpt22 -1.25000000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.75000000E-01 0.00000000E+00
-3.75000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 1.25000000E-01
-1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
kpt23 -1.25000000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.75000000E-01 0.00000000E+00
-3.75000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 1.25000000E-01
-1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
kpt24 -1.25000000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.75000000E-01 0.00000000E+00
-3.75000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 1.25000000E-01
-1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
kpt31 -8.33333333E-02 -1.66666667E-01 0.00000000E+00
-8.33333333E-02 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -2.50000000E-01 0.00000000E+00
-8.33333333E-02 -2.50000000E-01 8.33333333E-02
-8.33333333E-02 5.00000000E-01 0.00000000E+00
-1.66666667E-01 -4.16666667E-01 0.00000000E+00
-8.33333333E-02 -4.16666667E-01 8.33333333E-02
-2.50000000E-01 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -3.33333333E-01 8.33333333E-02
-8.33333333E-02 -3.33333333E-01 1.66666667E-01
-8.33333333E-02 3.33333333E-01 0.00000000E+00
-1.66666667E-01 4.16666667E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 0.00000000E+00
-1.66666667E-01 5.00000000E-01 8.33333333E-02
-3.33333333E-01 -4.16666667E-01 0.00000000E+00
-2.50000000E-01 -4.16666667E-01 8.33333333E-02
-1.66666667E-01 -4.16666667E-01 1.66666667E-01
-8.33333333E-02 -4.16666667E-01 2.50000000E-01
-8.33333333E-02 1.66666667E-01 0.00000000E+00
-1.66666667E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.33333333E-01 0.00000000E+00
-3.33333333E-01 4.16666667E-01 0.00000000E+00
-4.16666667E-01 5.00000000E-01 0.00000000E+00
-3.33333333E-01 5.00000000E-01 8.33333333E-02
-2.50000000E-01 5.00000000E-01 1.66666667E-01
-8.33333333E-02 0.00000000E+00 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
-4.16666667E-01 0.00000000E+00 0.00000000E+00
kpt32 -8.33333333E-02 -1.66666667E-01 0.00000000E+00
-8.33333333E-02 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -2.50000000E-01 0.00000000E+00
-8.33333333E-02 -2.50000000E-01 8.33333333E-02
-8.33333333E-02 5.00000000E-01 0.00000000E+00
-1.66666667E-01 -4.16666667E-01 0.00000000E+00
-8.33333333E-02 -4.16666667E-01 8.33333333E-02
-2.50000000E-01 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -3.33333333E-01 8.33333333E-02
-8.33333333E-02 -3.33333333E-01 1.66666667E-01
-8.33333333E-02 3.33333333E-01 0.00000000E+00
-1.66666667E-01 4.16666667E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 0.00000000E+00
-1.66666667E-01 5.00000000E-01 8.33333333E-02
-3.33333333E-01 -4.16666667E-01 0.00000000E+00
-2.50000000E-01 -4.16666667E-01 8.33333333E-02
-1.66666667E-01 -4.16666667E-01 1.66666667E-01
-8.33333333E-02 -4.16666667E-01 2.50000000E-01
-8.33333333E-02 1.66666667E-01 0.00000000E+00
-1.66666667E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.33333333E-01 0.00000000E+00
-3.33333333E-01 4.16666667E-01 0.00000000E+00
-4.16666667E-01 5.00000000E-01 0.00000000E+00
-3.33333333E-01 5.00000000E-01 8.33333333E-02
-2.50000000E-01 5.00000000E-01 1.66666667E-01
-8.33333333E-02 0.00000000E+00 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
-4.16666667E-01 0.00000000E+00 0.00000000E+00
kpt33 -8.33333333E-02 -1.66666667E-01 0.00000000E+00
-8.33333333E-02 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -2.50000000E-01 0.00000000E+00
-8.33333333E-02 -2.50000000E-01 8.33333333E-02
-8.33333333E-02 5.00000000E-01 0.00000000E+00
-1.66666667E-01 -4.16666667E-01 0.00000000E+00
-8.33333333E-02 -4.16666667E-01 8.33333333E-02
-2.50000000E-01 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -3.33333333E-01 8.33333333E-02
-8.33333333E-02 -3.33333333E-01 1.66666667E-01
-8.33333333E-02 3.33333333E-01 0.00000000E+00
-1.66666667E-01 4.16666667E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 0.00000000E+00
-1.66666667E-01 5.00000000E-01 8.33333333E-02
-3.33333333E-01 -4.16666667E-01 0.00000000E+00
-2.50000000E-01 -4.16666667E-01 8.33333333E-02
-1.66666667E-01 -4.16666667E-01 1.66666667E-01
-8.33333333E-02 -4.16666667E-01 2.50000000E-01
-8.33333333E-02 1.66666667E-01 0.00000000E+00
-1.66666667E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.33333333E-01 0.00000000E+00
-3.33333333E-01 4.16666667E-01 0.00000000E+00
-4.16666667E-01 5.00000000E-01 0.00000000E+00
-3.33333333E-01 5.00000000E-01 8.33333333E-02
-2.50000000E-01 5.00000000E-01 1.66666667E-01
-8.33333333E-02 0.00000000E+00 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
-4.16666667E-01 0.00000000E+00 0.00000000E+00
kpt34 -8.33333333E-02 -1.66666667E-01 0.00000000E+00
-8.33333333E-02 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -2.50000000E-01 0.00000000E+00
-8.33333333E-02 -2.50000000E-01 8.33333333E-02
-8.33333333E-02 5.00000000E-01 0.00000000E+00
-1.66666667E-01 -4.16666667E-01 0.00000000E+00
-8.33333333E-02 -4.16666667E-01 8.33333333E-02
-2.50000000E-01 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -3.33333333E-01 8.33333333E-02
-8.33333333E-02 -3.33333333E-01 1.66666667E-01
-8.33333333E-02 3.33333333E-01 0.00000000E+00
-1.66666667E-01 4.16666667E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 0.00000000E+00
-1.66666667E-01 5.00000000E-01 8.33333333E-02
-3.33333333E-01 -4.16666667E-01 0.00000000E+00
-2.50000000E-01 -4.16666667E-01 8.33333333E-02
-1.66666667E-01 -4.16666667E-01 1.66666667E-01
-8.33333333E-02 -4.16666667E-01 2.50000000E-01
-8.33333333E-02 1.66666667E-01 0.00000000E+00
-1.66666667E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.33333333E-01 0.00000000E+00
-3.33333333E-01 4.16666667E-01 0.00000000E+00
-4.16666667E-01 5.00000000E-01 0.00000000E+00
-3.33333333E-01 5.00000000E-01 8.33333333E-02
-2.50000000E-01 5.00000000E-01 1.66666667E-01
-8.33333333E-02 0.00000000E+00 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
-4.16666667E-01 0.00000000E+00 0.00000000E+00
kptrlatt11 2 -2 2 -2 2 2 -2 -2 2
kptrlatt12 2 -2 2 -2 2 2 -2 -2 2
kptrlatt13 2 -2 2 -2 2 2 -2 -2 2
kptrlatt14 2 -2 2 -2 2 2 -2 -2 2
kptrlatt21 4 -4 4 -4 4 4 -4 -4 4
kptrlatt22 4 -4 4 -4 4 4 -4 -4 4
kptrlatt23 4 -4 4 -4 4 4 -4 -4 4
kptrlatt24 4 -4 4 -4 4 4 -4 -4 4
kptrlatt31 6 -6 6 -6 6 6 -6 -6 6
kptrlatt32 6 -6 6 -6 6 6 -6 -6 6
kptrlatt33 6 -6 6 -6 6 6 -6 -6 6
kptrlatt34 6 -6 6 -6 6 6 -6 -6 6
kptrlen11 1.52000000E+01
kptrlen12 1.52000000E+01
kptrlen13 1.52000000E+01
kptrlen14 1.52000000E+01
kptrlen21 3.04000000E+01
kptrlen22 3.04000000E+01
kptrlen23 3.04000000E+01
kptrlen24 3.04000000E+01
kptrlen31 4.56000000E+01
kptrlen32 4.56000000E+01
kptrlen33 4.56000000E+01
kptrlen34 4.56000000E+01
P mkmem11 2
P mkmem12 2
P mkmem13 2
P mkmem14 2
P mkmem21 10
P mkmem22 10
P mkmem23 10
P mkmem24 10
P mkmem31 28
P mkmem32 28
P mkmem33 28
P mkmem34 28
natom 1
nband11 3
nband12 3
nband13 3
nband14 3
nband21 3
nband22 3
nband23 3
nband24 3
nband31 3
nband32 3
nband33 3
nband34 3
ndtset 12
ngfft 15 15 15
nkpt11 2
nkpt12 2
nkpt13 2
nkpt14 2
nkpt21 10
nkpt22 10
nkpt23 10
nkpt24 10
nkpt31 28
nkpt32 28
nkpt33 28
nkpt34 28
nstep 10
nsym 48
ntime 10
ntypat 1
occ11 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ12 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ13 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ14 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ21 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ22 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ23 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ24 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ31 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ32 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ33 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occ34 2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
2.000000 1.000000 0.000000
occopt 4
optcell 1
rprim 0.0000000000E+00 5.0000000000E-01 5.0000000000E-01
5.0000000000E-01 0.0000000000E+00 5.0000000000E-01
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
shiftk 5.00000000E-01 5.00000000E-01 5.00000000E-01
spgroup 225
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
toldfe 1.00000000E-06 Hartree
tsmear11 1.00000000E-02 Hartree
tsmear12 2.00000000E-02 Hartree
tsmear13 3.00000000E-02 Hartree
tsmear14 4.00000000E-02 Hartree
tsmear21 1.00000000E-02 Hartree
tsmear22 2.00000000E-02 Hartree
tsmear23 3.00000000E-02 Hartree
tsmear24 4.00000000E-02 Hartree
tsmear31 1.00000000E-02 Hartree
tsmear32 2.00000000E-02 Hartree
tsmear33 3.00000000E-02 Hartree
tsmear34 4.00000000E-02 Hartree
typat 1
wtk11 0.75000 0.25000
wtk12 0.75000 0.25000
wtk13 0.75000 0.25000
wtk14 0.75000 0.25000
wtk21 0.09375 0.09375 0.09375 0.18750 0.09375 0.09375
0.09375 0.18750 0.03125 0.03125
wtk22 0.09375 0.09375 0.09375 0.18750 0.09375 0.09375
0.09375 0.18750 0.03125 0.03125
wtk23 0.09375 0.09375 0.09375 0.18750 0.09375 0.09375
0.09375 0.18750 0.03125 0.03125
wtk24 0.09375 0.09375 0.09375 0.18750 0.09375 0.09375
0.09375 0.18750 0.03125 0.03125
wtk31 0.02778 0.02778 0.02778 0.05556 0.02778 0.02778
0.05556 0.02778 0.05556 0.05556 0.02778 0.02778
0.02778 0.05556 0.02778 0.05556 0.05556 0.05556
0.02778 0.02778 0.02778 0.02778 0.02778 0.05556
0.05556 0.00926 0.00926 0.00926
wtk32 0.02778 0.02778 0.02778 0.05556 0.02778 0.02778
0.05556 0.02778 0.05556 0.05556 0.02778 0.02778
0.02778 0.05556 0.02778 0.05556 0.05556 0.05556
0.02778 0.02778 0.02778 0.02778 0.02778 0.05556
0.05556 0.00926 0.00926 0.00926
wtk33 0.02778 0.02778 0.02778 0.05556 0.02778 0.02778
0.05556 0.02778 0.05556 0.05556 0.02778 0.02778
0.02778 0.05556 0.02778 0.05556 0.05556 0.05556
0.02778 0.02778 0.02778 0.02778 0.02778 0.05556
0.05556 0.00926 0.00926 0.00926
wtk34 0.02778 0.02778 0.02778 0.05556 0.02778 0.02778
0.05556 0.02778 0.05556 0.05556 0.02778 0.02778
0.02778 0.05556 0.02778 0.05556 0.05556 0.05556
0.02778 0.02778 0.02778 0.02778 0.02778 0.05556
0.05556 0.00926 0.00926 0.00926
znucl 13.00000
================================================================================
chkinp: Checking input parameters for consistency, jdtset= 11.
chkinp: Checking input parameters for consistency, jdtset= 12.
chkinp: Checking input parameters for consistency, jdtset= 13.
chkinp: Checking input parameters for consistency, jdtset= 14.
chkinp: Checking input parameters for consistency, jdtset= 21.
chkinp: Checking input parameters for consistency, jdtset= 22.
chkinp: Checking input parameters for consistency, jdtset= 23.
chkinp: Checking input parameters for consistency, jdtset= 24.
chkinp: Checking input parameters for consistency, jdtset= 31.
chkinp: Checking input parameters for consistency, jdtset= 32.
chkinp: Checking input parameters for consistency, jdtset= 33.
chkinp: Checking input parameters for consistency, jdtset= 34.
================================================================================
== DATASET 11 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 11, }
dimensions: {natom: 1, nkpt: 2, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 90, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--- Pseudopotential description ------------------------------------------------
- pspini: atom type 1 psp file is /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/Pspdir/Psdj_nc_sr_04_pw_std_psp8/Al.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/Pspdir/Psdj_nc_sr_04_pw_std_psp8/Al.psp8
- Al ONCVPSP-3.3.0 r_core= 1.76802 1.76802 1.70587
- 13.00000 3.00000 171102 znucl, zion, pspdat
8 -1012 2 4 600 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
5.99000000000000 5.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2 2
extension_switch 1
pspatm : epsatm= 0.57439192
--- l ekb(1:nproj) -->
0 5.725870 0.726131
1 6.190420 0.914022
2 -4.229503 -0.925599
pspatm: atomic psp has been read and splines computed
1.72317576E+00 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
_setup2: Arith. and geom. avg. npw (full set) are 89.750 89.749
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 11, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3515037024234 -2.352E+00 1.732E-03 2.558E-01
ETOT 2 -2.3516603712951 -1.567E-04 2.687E-08 1.111E-02
ETOT 3 -2.3516654661336 -5.095E-06 1.068E-07 3.747E-05
ETOT 4 -2.3516654898808 -2.375E-08 7.790E-10 1.373E-07
ETOT 5 -2.3516654899422 -6.135E-11 1.413E-12 1.561E-09
At SCF step 5, etot is converged :
for the second time, diff in etot= 6.135E-11 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.42712971E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.42712971E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.42712971E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 11, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -6.135E-11, res2: 1.561E-09, residm: 1.413E-12, diffor: null, }
etotal : -2.35166549E+00
entropy : 0.00000000E+00
fermie : 2.60581448E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -2.42712971E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -2.42712971E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -2.42712971E-06, ]
pressure_GPa: 7.1409E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93505925
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-2.42712970548859E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -2.42712970548861E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -2.42712970548861E-06
Total energy (etotal) [Ha]= -2.35166548994219E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 11, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3516655409507 -2.352E+00 4.202E-13 6.019E-08
ETOT 2 -2.3516655409654 -1.474E-11 7.554E-17 2.640E-09
ETOT 3 -2.3516655409665 -1.060E-12 1.999E-14 9.220E-12
At SCF step 3, etot is converged :
for the second time, diff in etot= 1.060E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.82598340E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.82598340E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.82598340E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 11, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8002767, 3.8002767, ]
- [ 3.8002767, 0.0000000, 3.8002767, ]
- [ 3.8002767, 3.8002767, 0.0000000, ]
lattice_lengths: [ 5.37440, 5.37440, 5.37440, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0976797E+02
convergence: {deltae: -1.060E-12, res2: 9.220E-12, residm: 1.999E-14, diffor: null, }
etotal : -2.35166554E+00
entropy : 0.00000000E+00
fermie : 2.60503390E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.82598340E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.82598340E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.82598340E-06, ]
pressure_GPa: 5.3722E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93517038
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60055338557285E+00 7.60055338557285E+00 7.60055338557285E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80027669278643E+00 3.80027669278643E+00
3.80027669278643E+00 0.00000000000000E+00 3.80027669278643E+00
3.80027669278643E+00 3.80027669278643E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09767974408601E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37440283970894E+00 5.37440283970894E+00 5.37440283970894E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.82598340114671E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.82598340114676E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.82598340114679E-06
Total energy (etotal) [Ha]= -2.35166554096648E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-5.10243E-08
Relative =-2.16971E-08
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 11, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3516656072399 -2.352E+00 1.637E-12 5.475E-07
ETOT 2 -2.3516656073737 -1.338E-10 2.510E-15 2.394E-08
ETOT 3 -2.3516656073833 -9.658E-12 1.813E-13 8.416E-11
At SCF step 3, etot is converged :
for the second time, diff in etot= 9.658E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.98822633E-10 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.98822633E-10 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.98822633E-10 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 11, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8011179, 3.8011179, ]
- [ 3.8011179, 0.0000000, 3.8011179, ]
- [ 3.8011179, 3.8011179, 0.0000000, ]
lattice_lengths: [ 5.37559, 5.37559, 5.37559, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0984088E+02
convergence: {deltae: -9.658E-12, res2: 8.416E-11, residm: 1.813E-13, diffor: null, }
etotal : -2.35166561E+00
entropy : 0.00000000E+00
fermie : 2.60266233E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -2.98822633E-10, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -2.98822633E-10, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -2.98822633E-10, ]
pressure_GPa: 8.7917E-06
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93550677
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60223577921863E+00 7.60223577921863E+00 7.60223577921863E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80111788960932E+00 3.80111788960932E+00
3.80111788960932E+00 0.00000000000000E+00 3.80111788960932E+00
3.80111788960932E+00 3.80111788960932E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09840882451185E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37559247166449E+00 5.37559247166449E+00 5.37559247166449E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-2.98822633168971E-10 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -2.98822633168971E-10 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -2.98822633168971E-10
Total energy (etotal) [Ha]= -2.35166560738332E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-6.64168E-08
Relative =-2.82425E-08
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 2.9882E-08 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 82.060E-15; max= 18.126E-14
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.602235779219 7.602235779219 7.602235779219 bohr
= 4.022929923545 4.022929923545 4.022929923545 angstroms
prteigrs : about to open file tbase4_3o_DS11_EIG
Fermi (or HOMO) energy (hartree) = 0.26027 Average Vxc (hartree)= -0.36714
Eigenvalues (hartree) for nkpt= 2 k points:
kpt# 1, nband= 3, wtk= 0.75000, kpt= -0.2500 0.5000 0.0000 (reduced coord)
0.09837 0.25745 0.42134
occupation numbers for kpt# 1
2.00000 1.33333 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 11, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.68012095515665E-01
hartree : 3.75185280131484E-03
xc : -1.11507667656370E+00
Ewald energy : -2.71392257918533E+00
psp_core : 1.56879271394294E-02
local_psp : 1.66253437235862E-01
non_local_psp : 4.25222444314949E-01
internal : -2.35007149874181E+00
'-kT*entropy' : -1.59410864151562E-03
total_energy : -2.35166560738332E+00
total_energy_eV : -6.39920811638971E+01
band_energy : 3.72533144177011E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.98822633E-10 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.98822633E-10 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.98822633E-10 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 8.7917E-06 GPa]
- sigma(1 1)= -8.79166540E-06 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -8.79166540E-06 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -8.79166540E-06 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 12 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 12, }
dimensions: {natom: 1, nkpt: 2, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 90, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 2.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 11.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_3o_DS11_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.750 89.749
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 12, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3532595976569 -2.353E+00 5.822E-12 2.716E-06
ETOT 2 -2.3532595985224 -8.655E-10 6.062E-16 1.330E-07
ETOT 3 -2.3532595985844 -6.198E-11 1.477E-12 3.421E-10
At SCF step 3, etot is converged :
for the second time, diff in etot= 6.198E-11 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.42771945E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.42771945E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.42771945E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 12, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -6.198E-11, res2: 3.421E-10, residm: 1.477E-12, diffor: null, }
etotal : -2.35325960E+00
entropy : 0.00000000E+00
fermie : 2.63400570E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -2.42771945E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -2.42771945E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -2.42771945E-06, ]
pressure_GPa: 7.1426E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93505931
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-2.42771945317580E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -2.42771945317585E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -2.42771945317582E-06
Total energy (etotal) [Ha]= -2.35325959858440E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 12, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3532596496030 -2.353E+00 3.345E-13 5.995E-08
ETOT 2 -2.3532596496176 -1.458E-11 2.661E-16 2.626E-09
ETOT 3 -2.3532596496186 -1.049E-12 1.986E-14 9.198E-12
At SCF step 3, etot is converged :
for the second time, diff in etot= 1.049E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.82583663E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.82583663E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.82583663E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 12, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8002768, 3.8002768, ]
- [ 3.8002768, 0.0000000, 3.8002768, ]
- [ 3.8002768, 3.8002768, 0.0000000, ]
lattice_lengths: [ 5.37440, 5.37440, 5.37440, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0976798E+02
convergence: {deltae: -1.049E-12, res2: 9.198E-12, residm: 1.986E-14, diffor: null, }
etotal : -2.35325965E+00
entropy : 0.00000000E+00
fermie : 2.63322590E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.82583663E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.82583663E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.82583663E-06, ]
pressure_GPa: 5.3718E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93517040
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60055352003532E+00 7.60055352003532E+00 7.60055352003532E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80027676001766E+00 3.80027676001766E+00
3.80027676001766E+00 0.00000000000000E+00 3.80027676001766E+00
3.80027676001766E+00 3.80027676001766E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09767980234364E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37440293478826E+00 5.37440293478826E+00 5.37440293478826E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.82583663472502E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.82583663472499E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.82583663472504E-06
Total energy (etotal) [Ha]= -2.35325964961863E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-5.10342E-08
Relative =-2.16866E-08
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 12, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3532597158818 -2.353E+00 3.393E-12 5.463E-07
ETOT 2 -2.3532597160152 -1.334E-10 2.206E-15 2.389E-08
ETOT 3 -2.3532597160249 -9.690E-12 1.809E-13 8.398E-11
At SCF step 3, etot is converged :
for the second time, diff in etot= 9.690E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.08177271E-09 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.08177271E-09 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.08177271E-09 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 12, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8011171, 3.8011171, ]
- [ 3.8011171, 0.0000000, 3.8011171, ]
- [ 3.8011171, 3.8011171, 0.0000000, ]
lattice_lengths: [ 5.37559, 5.37559, 5.37559, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0984081E+02
convergence: {deltae: -9.690E-12, res2: 8.398E-11, residm: 1.809E-13, diffor: null, }
etotal : -2.35325972E+00
entropy : 0.00000000E+00
fermie : 2.63085685E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -2.08177271E-09, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -2.08177271E-09, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -2.08177271E-09, ]
pressure_GPa: 6.1248E-05
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93550644
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60223412706635E+00 7.60223412706635E+00 7.60223412706635E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80111706353317E+00 3.80111706353317E+00
3.80111706353317E+00 0.00000000000000E+00 3.80111706353317E+00
3.80111706353317E+00 3.80111706353317E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09840810837847E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37559130341641E+00 5.37559130341641E+00 5.37559130341641E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-2.08177271474323E-09 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -2.08177271471613E-09 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -2.08177271471613E-09
Total energy (etotal) [Ha]= -2.35325971602487E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-6.64062E-08
Relative =-2.82188E-08
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 2.0818E-07 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 81.659E-15; max= 18.087E-14
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.602234127066 7.602234127066 7.602234127066 bohr
= 4.022929049263 4.022929049263 4.022929049263 angstroms
prteigrs : about to open file tbase4_3o_DS12_EIG
Fermi (or HOMO) energy (hartree) = 0.26309 Average Vxc (hartree)= -0.36714
Eigenvalues (hartree) for nkpt= 2 k points:
kpt# 1, nband= 3, wtk= 0.75000, kpt= -0.2500 0.5000 0.0000 (reduced coord)
0.09837 0.25745 0.42134
occupation numbers for kpt# 1
2.00000 1.33333 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 12, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.68012449801914E-01
hartree : 3.75185539515713E-03
xc : -1.11507685043994E+00
Ewald energy : -2.71392316898741E+00
psp_core : 1.56879373675478E-02
local_psp : 1.66253754090104E-01
non_local_psp : 4.25222524030791E-01
internal : -2.35007149874183E+00
'-kT*entropy' : -3.18821728303177E-03
total_energy : -2.35325971602487E+00
total_energy_eV : -6.40354590698602E+01
band_energy : 3.72533683822830E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.08177271E-09 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.08177271E-09 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.08177271E-09 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 6.1248E-05 GPa]
- sigma(1 1)= -6.12478678E-05 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -6.12478678E-05 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -6.12478678E-05 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 13 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 13, }
dimensions: {natom: 1, nkpt: 2, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 90, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 3.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 12.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_3o_DS12_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.750 89.749
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 13, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3548537063178 -2.355E+00 5.722E-12 2.712E-06
ETOT 2 -2.3548537071820 -8.643E-10 4.349E-15 1.328E-07
ETOT 3 -2.3548537072439 -6.188E-11 1.474E-12 3.416E-10
At SCF step 3, etot is converged :
for the second time, diff in etot= 6.188E-11 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.42772127E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.42772127E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.42772127E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 13, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -6.188E-11, res2: 3.416E-10, residm: 1.474E-12, diffor: null, }
etotal : -2.35485371E+00
entropy : 0.00000000E+00
fermie : 2.66219789E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -2.42772127E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -2.42772127E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -2.42772127E-06, ]
pressure_GPa: 7.1426E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93505931
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-2.42772127325851E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -2.42772127325845E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -2.42772127325851E-06
Total energy (etotal) [Ha]= -2.35485370724392E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 13, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3548537582627 -2.355E+00 3.662E-13 5.995E-08
ETOT 2 -2.3548537582772 -1.456E-11 2.310E-16 2.626E-09
ETOT 3 -2.3548537582783 -1.044E-12 1.986E-14 9.198E-12
At SCF step 3, etot is converged :
for the second time, diff in etot= 1.044E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.82583604E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.82583604E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.82583604E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 13, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8002768, 3.8002768, ]
- [ 3.8002768, 0.0000000, 3.8002768, ]
- [ 3.8002768, 3.8002768, 0.0000000, ]
lattice_lengths: [ 5.37440, 5.37440, 5.37440, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0976798E+02
convergence: {deltae: -1.044E-12, res2: 9.198E-12, residm: 1.986E-14, diffor: null, }
etotal : -2.35485376E+00
entropy : 0.00000000E+00
fermie : 2.66141809E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.82583604E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.82583604E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.82583604E-06, ]
pressure_GPa: 5.3718E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93517040
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60055352045030E+00 7.60055352045030E+00 7.60055352045030E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80027676022515E+00 3.80027676022515E+00
3.80027676022515E+00 0.00000000000000E+00 3.80027676022515E+00
3.80027676022515E+00 3.80027676022515E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09767980252343E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37440293508170E+00 5.37440293508170E+00 5.37440293508170E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.82583603995450E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.82583603995455E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.82583603995461E-06
Total energy (etotal) [Ha]= -2.35485375827826E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-5.10343E-08
Relative =-2.16720E-08
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 13, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3548538245418 -2.355E+00 2.710E-12 5.463E-07
ETOT 2 -2.3548538246752 -1.334E-10 2.452E-15 2.389E-08
ETOT 3 -2.3548538246848 -9.674E-12 1.809E-13 8.398E-11
At SCF step 3, etot is converged :
for the second time, diff in etot= 9.674E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.08769281E-09 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.08769281E-09 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.08769281E-09 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 13, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8011171, 3.8011171, ]
- [ 3.8011171, 0.0000000, 3.8011171, ]
- [ 3.8011171, 3.8011171, 0.0000000, ]
lattice_lengths: [ 5.37559, 5.37559, 5.37559, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0984081E+02
convergence: {deltae: -9.674E-12, res2: 8.398E-11, residm: 1.809E-13, diffor: null, }
etotal : -2.35485382E+00
entropy : 0.00000000E+00
fermie : 2.65904905E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -2.08769281E-09, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -2.08769281E-09, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -2.08769281E-09, ]
pressure_GPa: 6.1422E-05
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93550644
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60223412144730E+00 7.60223412144730E+00 7.60223412144730E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80111706072365E+00 3.80111706072365E+00
3.80111706072365E+00 0.00000000000000E+00 3.80111706072365E+00
3.80111706072365E+00 3.80111706072365E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09840810594287E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37559129944314E+00 5.37559129944314E+00 5.37559129944314E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-2.08769281072677E-09 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -2.08769281069966E-09 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -2.08769281069966E-09
Total energy (etotal) [Ha]= -2.35485382468483E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-6.64066E-08
Relative =-2.81999E-08
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 2.0877E-07 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 81.728E-15; max= 18.087E-14
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.602234121447 7.602234121447 7.602234121447 bohr
= 4.022929046290 4.022929046290 4.022929046290 angstroms
prteigrs : about to open file tbase4_3o_DS13_EIG
Fermi (or HOMO) energy (hartree) = 0.26590 Average Vxc (hartree)= -0.36714
Eigenvalues (hartree) for nkpt= 2 k points:
kpt# 1, nband= 3, wtk= 0.75000, kpt= -0.2500 0.5000 0.0000 (reduced coord)
0.09837 0.25745 0.42134
occupation numbers for kpt# 1
2.00000 1.33333 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 13, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.68012450987686E-01
hartree : 3.75185540386304E-03
xc : -1.11507685101523E+00
Ewald energy : -2.71392317099334E+00
psp_core : 1.56879374023341E-02
local_psp : 1.66253755165386E-01
non_local_psp : 4.25222524296083E-01
internal : -2.35007149875322E+00
'-kT*entropy' : -4.78232593160563E-03
total_energy : -2.35485382468483E+00
total_energy_eV : -6.40788369763246E+01
band_energy : 3.72533685651108E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.08769281E-09 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.08769281E-09 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.08769281E-09 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 6.1422E-05 GPa]
- sigma(1 1)= -6.14220431E-05 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -6.14220431E-05 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -6.14220431E-05 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 14 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 14, }
dimensions: {natom: 1, nkpt: 2, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 90, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 4.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 13.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_3o_DS13_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.750 89.749
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 14, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3564478254426 -2.356E+00 5.830E-12 2.712E-06
ETOT 2 -2.3564478263069 -8.643E-10 5.386E-16 1.328E-07
ETOT 3 -2.3564478263687 -6.187E-11 1.474E-12 3.416E-10
At SCF step 3, etot is converged :
for the second time, diff in etot= 6.187E-11 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.42943142E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.42943142E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.42943142E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 14, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -6.187E-11, res2: 3.416E-10, residm: 1.474E-12, diffor: null, }
etotal : -2.35644783E+00
entropy : 0.00000000E+00
fermie : 2.69038947E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -2.42943142E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -2.42943142E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -2.42943142E-06, ]
pressure_GPa: 7.1476E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93505934
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-2.42943142244616E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -2.42943142244619E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -2.42943142244616E-06
Total energy (etotal) [Ha]= -2.35644782636874E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 14, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3564478774593 -2.356E+00 2.876E-13 6.004E-08
ETOT 2 -2.3564478774739 -1.460E-11 2.687E-16 2.629E-09
ETOT 3 -2.3564478774749 -1.050E-12 1.989E-14 9.211E-12
At SCF step 3, etot is converged :
for the second time, diff in etot= 1.050E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.82712719E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.82712719E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.82712719E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 14, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8002770, 3.8002770, ]
- [ 3.8002770, 0.0000000, 3.8002770, ]
- [ 3.8002770, 3.8002770, 0.0000000, ]
lattice_lengths: [ 5.37440, 5.37440, 5.37440, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0976800E+02
convergence: {deltae: -1.050E-12, res2: 9.211E-12, residm: 1.989E-14, diffor: null, }
etotal : -2.35644788E+00
entropy : 0.00000000E+00
fermie : 2.68960911E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.82712719E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.82712719E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.82712719E-06, ]
pressure_GPa: 5.3756E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93517052
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60055391036432E+00 7.60055391036432E+00 7.60055391036432E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80027695518216E+00 3.80027695518216E+00
3.80027695518216E+00 0.00000000000000E+00 3.80027695518216E+00
3.80027695518216E+00 3.80027695518216E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09767997145880E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37440321079254E+00 5.37440321079254E+00 5.37440321079254E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.82712718743857E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.82712718743854E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.82712718743854E-06
Total energy (etotal) [Ha]= -2.35644787747492E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-5.11062E-08
Relative =-2.16878E-08
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 14, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3564479438327 -2.356E+00 3.407E-12 5.471E-07
ETOT 2 -2.3564479439664 -1.337E-10 2.032E-15 2.392E-08
ETOT 3 -2.3564479439760 -9.645E-12 1.811E-13 8.410E-11
At SCF step 3, etot is converged :
for the second time, diff in etot= 9.645E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.10276955E-09 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.10276955E-09 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.10276955E-09 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 14, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8011179, 3.8011179, ]
- [ 3.8011179, 0.0000000, 3.8011179, ]
- [ 3.8011179, 3.8011179, 0.0000000, ]
lattice_lengths: [ 5.37559, 5.37559, 5.37559, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0984088E+02
convergence: {deltae: -9.645E-12, res2: 8.410E-11, residm: 1.811E-13, diffor: null, }
etotal : -2.35644794E+00
entropy : 0.00000000E+00
fermie : 2.68723837E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -2.10276955E-09, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -2.10276955E-09, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -2.10276955E-09, ]
pressure_GPa: 6.1866E-05
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93550680
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60223571477244E+00 7.60223571477244E+00 7.60223571477244E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80111785738622E+00 3.80111785738622E+00
3.80111785738622E+00 0.00000000000000E+00 3.80111785738622E+00
3.80111785738622E+00 3.80111785738622E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09840879657733E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37559242609415E+00 5.37559242609415E+00 5.37559242609415E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-2.10276954930397E-09 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -2.10276954930397E-09 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -2.10276954935818E-09
Total energy (etotal) [Ha]= -2.35644794397605E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-6.65011E-08
Relative =-2.82209E-08
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 2.1028E-07 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 81.743E-15; max= 18.113E-14
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.602235714772 7.602235714772 7.602235714772 bohr
= 4.022929889441 4.022929889441 4.022929889441 angstroms
prteigrs : about to open file tbase4_3o_DS14_EIG
Fermi (or HOMO) energy (hartree) = 0.26872 Average Vxc (hartree)= -0.36714
Eigenvalues (hartree) for nkpt= 2 k points:
kpt# 1, nband= 3, wtk= 0.75000, kpt= -0.2500 0.5000 0.0000 (reduced coord)
0.09837 0.25745 0.42134
occupation numbers for kpt# 1
2.00000 1.33333 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 14, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.68012435875115E-01
hartree : 3.75185171627605E-03
xc : -1.11507668497712E+00
Ewald energy : -2.71392260219199E+00
psp_core : 1.56879275384017E-02
local_psp : 1.66253422534841E-01
non_local_psp : 4.25222445210670E-01
internal : -2.35007120429381E+00
'-kT*entropy' : -6.37673968224301E-03
total_energy : -2.35644794397605E+00
total_energy_eV : -6.41222151720801E+01
band_energy : 3.72533459620834E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -2.10276955E-09 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -2.10276955E-09 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.10276955E-09 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 6.1866E-05 GPa]
- sigma(1 1)= -6.18656160E-05 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -6.18656160E-05 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -6.18656160E-05 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 21 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 21, }
dimensions: {natom: 1, nkpt: 10, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 92, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 14.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_3o_DS14_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.000 88.972
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 21, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3567926034953 -2.357E+00 5.696E-01 2.322E-04
ETOT 2 -2.3567926735839 -7.009E-08 1.146E-07 8.838E-06
ETOT 3 -2.3567926773909 -3.807E-09 4.434E-09 4.287E-08
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.807E-09 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 4.09517297E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 4.09517297E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.09517297E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 21, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -3.807E-09, res2: 4.287E-08, residm: 4.434E-09, diffor: null, }
etotal : -2.35679268E+00
entropy : 0.00000000E+00
fermie : 2.91822887E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 4.09517297E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 4.09517297E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 4.09517297E-05, ]
pressure_GPa: -1.2048E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93036975
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
4.09517296586539E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 4.09517296586539E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 4.09517296586538E-05
Total energy (etotal) [Ha]= -2.35679267739093E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 21, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3568072812211 -2.357E+00 6.012E-10 1.785E-05
ETOT 2 -2.3568072857890 -4.568E-09 6.288E-13 7.675E-07
ETOT 3 -2.3568072861065 -3.175E-10 1.379E-11 2.725E-09
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.175E-10 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 3.14091507E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 3.14091507E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.14091507E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 21, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7953315, 3.7953315, ]
- [ 3.7953315, 0.0000000, 3.7953315, ]
- [ 3.7953315, 3.7953315, 0.0000000, ]
lattice_lengths: [ 5.36741, 5.36741, 5.36741, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0934002E+02
convergence: {deltae: -3.175E-10, res2: 2.725E-09, residm: 1.379E-11, diffor: null, }
etotal : -2.35680729E+00
entropy : 0.00000000E+00
fermie : 2.93214568E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 3.14091507E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 3.14091507E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.14091507E-05, ]
pressure_GPa: -9.2409E-01
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92848373
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.59066300563783E+00 7.59066300563783E+00 7.59066300563783E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.79533150281891E+00 3.79533150281891E+00
3.79533150281891E+00 0.00000000000000E+00 3.79533150281891E+00
3.79533150281891E+00 3.79533150281891E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09340018123675E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.36740928498837E+00 5.36740928498837E+00 5.36740928498837E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
3.14091507464306E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.14091507464305E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 3.14091507464304E-05
Total energy (etotal) [Ha]= -2.35680728610651E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.46087E-05
Relative =-6.19854E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 21, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3568282138804 -2.357E+00 6.011E-09 1.866E-04
ETOT 2 -2.3568282605883 -4.671E-08 2.541E-12 7.963E-06
ETOT 3 -2.3568282638087 -3.220E-09 1.386E-10 2.810E-08
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.220E-09 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.09643255E-07 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.09643255E-07 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.09643255E-07 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 21, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7802051, 3.7802051, ]
- [ 3.7802051, 0.0000000, 3.7802051, ]
- [ 3.7802051, 3.7802051, 0.0000000, ]
lattice_lengths: [ 5.34602, 5.34602, 5.34602, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0803789E+02
convergence: {deltae: -3.220E-09, res2: 2.810E-08, residm: 1.386E-10, diffor: null, }
etotal : -2.35682826E+00
entropy : 0.00000000E+00
fermie : 2.97758813E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 2.09643255E-07, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 2.09643255E-07, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 2.09643255E-07, ]
pressure_GPa: -6.1679E-03
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92240406
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.56041021448918E+00 7.56041021448918E+00 7.56041021448918E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.78020510724459E+00 3.78020510724459E+00
3.78020510724459E+00 0.00000000000000E+00 3.78020510724459E+00
3.78020510724459E+00 3.78020510724459E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.08037888880263E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.34601733121734E+00 5.34601733121734E+00 5.34601733121734E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
2.09643255382348E-07 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 2.09643255382266E-07 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 2.09643255382321E-07
Total energy (etotal) [Ha]= -2.35682826380875E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.09777E-05
Relative =-8.90086E-06
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 2.0964E-05 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 40.644E-12; max= 13.857E-11
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.560410214489 7.560410214489 7.560410214489 bohr
= 4.000796787872 4.000796787872 4.000796787872 angstroms
prteigrs : about to open file tbase4_3o_DS21_EIG
Fermi (or HOMO) energy (hartree) = 0.29776 Average Vxc (hartree)= -0.36916
Eigenvalues (hartree) for nkpt= 10 k points:
kpt# 1, nband= 3, wtk= 0.09375, kpt= -0.1250 -0.2500 0.0000 (reduced coord)
-0.06746 0.49135 0.62367
occupation numbers for kpt# 1
2.00000 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 21, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.70608408370381E-01
hartree : 4.16476399822845E-03
xc : -1.11942858234759E+00
Ewald energy : -2.72893649262206E+00
psp_core : 1.59497355852129E-02
local_psp : 1.77625774099153E-01
non_local_psp : 4.23742164113258E-01
internal : -2.35627422880342E+00
'-kT*entropy' : -5.54035005331959E-04
total_energy : -2.35682826380875E+00
total_energy_eV : -6.41325642019447E+01
band_energy : 3.80198462555459E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.09643255E-07 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.09643255E-07 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.09643255E-07 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= -6.1679E-03 GPa]
- sigma(1 1)= 6.16791752E-03 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= 6.16791752E-03 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= 6.16791752E-03 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 22 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 22, }
dimensions: {natom: 1, nkpt: 10, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 92, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 2.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 21.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_3o_DS21_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.000 88.972
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 22, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3573775902248 -2.357E+00 1.033E-08 8.629E-04
ETOT 2 -2.3573778634097 -2.732E-07 2.651E-11 4.218E-05
ETOT 3 -2.3573778830406 -1.963E-08 1.201E-09 1.126E-07
At SCF step 3, etot is converged :
for the second time, diff in etot= 1.963E-08 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 4.05317570E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 4.05317570E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.05317570E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 22, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -1.963E-08, res2: 1.126E-07, residm: 1.201E-09, diffor: null, }
etotal : -2.35737788E+00
entropy : 0.00000000E+00
fermie : 2.90874215E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 4.05317570E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 4.05317570E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 4.05317570E-05, ]
pressure_GPa: -1.1925E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93046598
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
4.05317570014033E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 4.05317570014034E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 4.05317570014033E-05
Total energy (etotal) [Ha]= -2.35737788304055E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 22, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3573922187401 -2.357E+00 5.812E-10 1.809E-05
ETOT 2 -2.3573922233120 -4.572E-09 2.767E-13 7.818E-07
ETOT 3 -2.3573922236341 -3.221E-10 1.446E-11 2.779E-09
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.221E-10 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 3.11507035E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 3.11507035E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.11507035E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 22, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7953794, 3.7953794, ]
- [ 3.7953794, 0.0000000, 3.7953794, ]
- [ 3.7953794, 3.7953794, 0.0000000, ]
lattice_lengths: [ 5.36748, 5.36748, 5.36748, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0934416E+02
convergence: {deltae: -3.221E-10, res2: 2.779E-09, residm: 1.446E-11, diffor: null, }
etotal : -2.35739222E+00
entropy : 0.00000000E+00
fermie : 2.92263412E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 3.11507035E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 3.11507035E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.11507035E-05, ]
pressure_GPa: -9.1649E-01
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92857562
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.59075875940368E+00 7.59075875940368E+00 7.59075875940368E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.79537937970184E+00 3.79537937970184E+00
3.79537937970184E+00 0.00000000000000E+00 3.79537937970184E+00
3.79537937970184E+00 3.79537937970184E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09344156043074E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.36747699312553E+00 5.36747699312553E+00 5.36747699312553E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
3.11507034823932E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.11507034823933E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 3.11507034823932E-05
Total energy (etotal) [Ha]= -2.35739222363410E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.43406E-05
Relative =-6.08326E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 22, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3574127854749 -2.357E+00 6.003E-09 1.887E-04
ETOT 2 -2.3574128322059 -4.673E-08 1.923E-12 8.084E-06
ETOT 3 -2.3574128354652 -3.259E-09 1.437E-10 2.861E-08
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.259E-09 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -3.07861898E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -3.07861898E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -3.07861898E-08 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 22, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7802748, 3.7802748, ]
- [ 3.7802748, 0.0000000, 3.7802748, ]
- [ 3.7802748, 3.7802748, 0.0000000, ]
lattice_lengths: [ 5.34612, 5.34612, 5.34612, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0804386E+02
convergence: {deltae: -3.259E-09, res2: 2.861E-08, residm: 1.437E-10, diffor: null, }
etotal : -2.35741284E+00
entropy : 0.00000000E+00
fermie : 2.96845047E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -3.07861898E-08, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -3.07861898E-08, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -3.07861898E-08, ]
pressure_GPa: 9.0576E-04
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92247099
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.56054960292104E+00 7.56054960292104E+00 7.56054960292104E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.78027480146052E+00 3.78027480146052E+00
3.78027480146052E+00 0.00000000000000E+00 3.78027480146052E+00
3.78027480146052E+00 3.78027480146052E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.08043864551872E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.34611589372272E+00 5.34611589372272E+00 5.34611589372272E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-3.07861897908251E-08 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -3.07861897908251E-08 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -3.07861897908522E-08
Total energy (etotal) [Ha]= -2.35741283546517E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.06118E-05
Relative =-8.74345E-06
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 3.0786E-06 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 41.449E-12; max= 14.373E-11
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.560549602921 7.560549602921 7.560549602921 bohr
= 4.000870549053 4.000870549053 4.000870549053 angstroms
prteigrs : about to open file tbase4_3o_DS22_EIG
Fermi (or HOMO) energy (hartree) = 0.29685 Average Vxc (hartree)= -0.36917
Eigenvalues (hartree) for nkpt= 10 k points:
kpt# 1, nband= 3, wtk= 0.09375, kpt= -0.1250 -0.2500 0.0000 (reduced coord)
-0.06748 0.49131 0.62362
occupation numbers for kpt# 1
2.00000 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 22, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.70211217695470E-01
hartree : 4.10251576254320E-03
xc : -1.11937135379182E+00
Ewald energy : -2.72888618117669E+00
psp_core : 1.59488534399612E-02
local_psp : 1.77646836373078E-01
non_local_psp : 4.23912316732243E-01
internal : -2.35643579496521E+00
'-kT*entropy' : -9.77040499959224E-04
total_energy : -2.35741283546517E+00
total_energy_eV : -6.41484712070760E+01
band_energy : 3.79947173170949E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -3.07861898E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -3.07861898E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -3.07861898E-08 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 9.0576E-04 GPa]
- sigma(1 1)= -9.05760975E-04 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -9.05760975E-04 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -9.05760975E-04 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 23 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 23, }
dimensions: {natom: 1, nkpt: 10, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 92, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 3.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 22.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_3o_DS22_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.000 88.972
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 23, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3576346208880 -2.358E+00 1.016E-08 8.586E-04
ETOT 2 -2.3576348933859 -2.725E-07 2.643E-11 4.191E-05
ETOT 3 -2.3576349129454 -1.956E-08 1.194E-09 1.117E-07
At SCF step 3, etot is converged :
for the second time, diff in etot= 1.956E-08 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 4.45370999E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 4.45370999E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.45370999E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 23, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -1.956E-08, res2: 1.117E-07, residm: 1.194E-09, diffor: null, }
etotal : -2.35763491E+00
entropy : 0.00000000E+00
fermie : 2.88055570E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 4.45370999E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 4.45370999E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 4.45370999E-05, ]
pressure_GPa: -1.3103E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93040268
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
4.45370998762694E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 4.45370998762693E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 4.45370998762694E-05
Total energy (etotal) [Ha]= -2.35763491294544E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 23, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3576522280036 -2.358E+00 6.978E-10 2.199E-05
ETOT 2 -2.3576522335277 -5.524E-09 2.967E-13 9.516E-07
ETOT 3 -2.3576522339186 -3.909E-10 1.775E-11 3.372E-09
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.909E-10 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 3.42766987E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 3.42766987E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.42766987E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 23, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7949228, 3.7949228, ]
- [ 3.7949228, 0.0000000, 3.7949228, ]
- [ 3.7949228, 3.7949228, 0.0000000, ]
lattice_lengths: [ 5.36683, 5.36683, 5.36683, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0930470E+02
convergence: {deltae: -3.909E-10, res2: 3.372E-09, residm: 1.775E-11, diffor: null, }
etotal : -2.35765223E+00
entropy : 0.00000000E+00
fermie : 2.89576335E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 3.42766987E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 3.42766987E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.42766987E-05, ]
pressure_GPa: -1.0085E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92832097
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.58984554122821E+00 7.58984554122821E+00 7.58984554122821E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.79492277061411E+00 3.79492277061411E+00
3.79492277061411E+00 0.00000000000000E+00 3.79492277061411E+00
3.79492277061411E+00 3.79492277061411E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09304696328530E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.36683125036095E+00 5.36683125036095E+00 5.36683125036095E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
3.42766987373124E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.42766987373126E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 3.42766987373125E-05
Total energy (etotal) [Ha]= -2.35765223391858E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.73210E-05
Relative =-7.34673E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 23, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3576771266726 -2.358E+00 7.320E-09 2.314E-04
ETOT 2 -2.3576771834639 -5.679E-08 3.060E-12 9.920E-06
ETOT 3 -2.3576771874432 -3.979E-09 1.777E-10 3.498E-08
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.979E-09 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.55308507E-07 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.55308507E-07 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.55308507E-07 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 23, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7782522, 3.7782522, ]
- [ 3.7782522, 0.0000000, 3.7782522, ]
- [ 3.7782522, 3.7782522, 0.0000000, ]
lattice_lengths: [ 5.34326, 5.34326, 5.34326, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0787053E+02
convergence: {deltae: -3.979E-09, res2: 3.498E-08, residm: 1.777E-10, diffor: null, }
etotal : -2.35767719E+00
entropy : 0.00000000E+00
fermie : 2.94620123E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.55308507E-07, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.55308507E-07, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.55308507E-07, ]
pressure_GPa: 4.5693E-03
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92157081
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.55650441471859E+00 7.55650441471859E+00 7.55650441471859E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77825220735930E+00 3.77825220735930E+00
3.77825220735930E+00 0.00000000000000E+00 3.77825220735930E+00
3.77825220735930E+00 3.77825220735930E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07870534309507E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.34325551371360E+00 5.34325551371360E+00 5.34325551371360E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.55308506949426E-07 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.55308506949480E-07 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.55308506949480E-07
Total energy (etotal) [Ha]= -2.35767718744323E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.49535E-05
Relative =-1.05840E-05
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 1.5531E-05 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 50.937E-12; max= 17.771E-11
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.556504414719 7.556504414719 7.556504414719 bohr
= 3.998729927644 3.998729927644 3.998729927644 angstroms
prteigrs : about to open file tbase4_3o_DS23_EIG
Fermi (or HOMO) energy (hartree) = 0.29462 Average Vxc (hartree)= -0.36937
Eigenvalues (hartree) for nkpt= 10 k points:
kpt# 1, nband= 3, wtk= 0.09375, kpt= -0.1250 -0.2500 0.0000 (reduced coord)
-0.06723 0.49209 0.62460
occupation numbers for kpt# 1
2.00000 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 23, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.70085438146142E-01
hartree : 4.08977255726042E-03
xc : -1.11978239855001E+00
Ewald energy : -2.73034702306603E+00
psp_core : 1.59744806295350E-02
local_psp : 1.78523473595674E-01
non_local_psp : 4.24194520986737E-01
internal : -2.35726173570069E+00
'-kT*entropy' : -4.15451742539138E-04
total_energy : -2.35767718744323E+00
total_energy_eV : -6.41556645908558E+01
band_energy : 3.80441908880963E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.55308507E-07 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.55308507E-07 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.55308507E-07 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 4.5693E-03 GPa]
- sigma(1 1)= -4.56933403E-03 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -4.56933403E-03 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -4.56933403E-03 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 24 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 24, }
dimensions: {natom: 1, nkpt: 10, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 92, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 4.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 23.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_3o_DS23_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.000 88.972
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 24, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3578212274627 -2.358E+00 1.231E-08 1.044E-03
ETOT 2 -2.3578215596562 -3.322E-07 3.250E-11 5.091E-05
ETOT 3 -2.3578215834715 -2.382E-08 1.450E-09 1.354E-07
At SCF step 3, etot is converged :
for the second time, diff in etot= 2.382E-08 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 4.15235329E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 4.15235329E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.15235329E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 24, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -2.382E-08, res2: 1.354E-07, residm: 1.450E-09, diffor: null, }
etotal : -2.35782158E+00
entropy : 0.00000000E+00
fermie : 2.87562393E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 4.15235329E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 4.15235329E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 4.15235329E-05, ]
pressure_GPa: -1.2217E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93052862
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
4.15235329276296E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 4.15235329276295E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 4.15235329276295E-05
Total energy (etotal) [Ha]= -2.35782158347152E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 24, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3578366423806 -2.358E+00 6.087E-10 1.925E-05
ETOT 2 -2.3578366472014 -4.821E-09 1.518E-13 8.345E-07
ETOT 3 -2.3578366475439 -3.425E-10 1.569E-11 2.954E-09
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.425E-10 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 3.19760217E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 3.19760217E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.19760217E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 24, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7952663, 3.7952663, ]
- [ 3.7952663, 0.0000000, 3.7952663, ]
- [ 3.7952663, 3.7952663, 0.0000000, ]
lattice_lengths: [ 5.36732, 5.36732, 5.36732, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0933438E+02
convergence: {deltae: -3.425E-10, res2: 2.954E-09, residm: 1.569E-11, diffor: null, }
etotal : -2.35783665E+00
entropy : 0.00000000E+00
fermie : 2.88962878E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 3.19760217E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 3.19760217E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.19760217E-05, ]
pressure_GPa: -9.4077E-01
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92858204
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.59053263449250E+00 7.59053263449250E+00 7.59053263449250E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.79526631724625E+00 3.79526631724625E+00
3.79526631724625E+00 0.00000000000000E+00 3.79526631724625E+00
3.79526631724625E+00 3.79526631724625E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09334384410828E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.36731709866744E+00 5.36731709866744E+00 5.36731709866744E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
3.19760217047759E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.19760217047759E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 3.19760217047759E-05
Total energy (etotal) [Ha]= -2.35783664754389E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.50641E-05
Relative =-6.38896E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 24, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3578584233619 -2.358E+00 6.411E-09 2.039E-04
ETOT 2 -2.3578584733084 -4.995E-08 2.432E-12 8.758E-06
ETOT 3 -2.3578584768245 -3.516E-09 1.582E-10 3.087E-08
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.516E-09 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -6.54501924E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -6.54501924E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -6.54501924E-08 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 24, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7796667, 3.7796667, ]
- [ 3.7796667, 0.0000000, 3.7796667, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
lattice_lengths: [ 5.34526, 5.34526, 5.34526, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0799173E+02
convergence: {deltae: -3.516E-09, res2: 3.087E-08, residm: 1.582E-10, diffor: null, }
etotal : -2.35785848E+00
entropy : 0.00000000E+00
fermie : 2.93622147E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -6.54501924E-08, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -6.54501924E-08, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -6.54501924E-08, ]
pressure_GPa: 1.9256E-03
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92224738
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.55933338856807E+00 7.55933338856807E+00 7.55933338856807E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77966669428403E+00 3.77966669428403E+00
3.77966669428403E+00 0.00000000000000E+00 3.77966669428403E+00
3.77966669428403E+00 3.77966669428403E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07991732087156E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.34525590030636E+00 5.34525590030636E+00 5.34525590030636E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-6.54501923512190E-08 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -6.54501923512732E-08 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -6.54501923512461E-08
Total energy (etotal) [Ha]= -2.35785847682446E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.18293E-05
Relative =-9.25814E-06
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 6.5450E-06 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 45.347E-12; max= 15.820E-11
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.559333388568 7.559333388568 7.559333388568 bohr
= 4.000226956135 4.000226956135 4.000226956135 angstroms
prteigrs : about to open file tbase4_3o_DS24_EIG
Fermi (or HOMO) energy (hartree) = 0.29362 Average Vxc (hartree)= -0.36923
Eigenvalues (hartree) for nkpt= 10 k points:
kpt# 1, nband= 3, wtk= 0.09375, kpt= -0.1250 -0.2500 0.0000 (reduced coord)
-0.06741 0.49153 0.62391
occupation numbers for kpt# 1
2.00000 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 24, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.69846028588563E-01
hartree : 4.06067320060200E-03
xc : -1.11947069063319E+00
Ewald energy : -2.72932522922108E+00
psp_core : 1.59565526686256E-02
local_psp : 1.77920286288706E-01
non_local_psp : 4.24136117022590E-01
internal : -2.35687626208519E+00
'-kT*entropy' : -9.82214739272263E-04
total_energy : -2.35785847682446E+00
total_energy_eV : -6.41605977262308E+01
band_energy : 3.79877449274362E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -6.54501924E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -6.54501924E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -6.54501924E-08 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 1.9256E-03 GPa]
- sigma(1 1)= -1.92561114E-03 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -1.92561114E-03 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -1.92561114E-03 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 31 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 31, }
dimensions: {natom: 1, nkpt: 28, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 94, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 24.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_3o_DS24_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.361 89.328
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 31, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581455628763 -2.358E+00 1.505E-03 4.369E-04
ETOT 2 -2.3581457592292 -1.964E-07 4.010E-07 2.184E-05
ETOT 3 -2.3581457716959 -1.247E-08 6.001E-08 3.401E-08
At SCF step 3, etot is converged :
for the second time, diff in etot= 1.247E-08 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 5.59204431E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 5.59204431E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 5.59204431E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 31, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -1.247E-08, res2: 3.401E-08, residm: 6.001E-08, diffor: null, }
etotal : -2.35814577E+00
entropy : 0.00000000E+00
fermie : 2.82286622E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 5.59204431E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 5.59204431E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 5.59204431E-05, ]
pressure_GPa: -1.6452E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92998653
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
5.59204431412555E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 5.59204431412555E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 5.59204431412553E-05
Total energy (etotal) [Ha]= -2.35814577169586E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 31, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581729691345 -2.358E+00 2.501E-08 3.498E-05
ETOT 2 -2.3581729777672 -8.633E-09 6.441E-11 1.525E-06
ETOT 3 -2.3581729783887 -6.214E-10 5.184E-11 5.340E-09
At SCF step 3, etot is converged :
for the second time, diff in etot= 6.214E-10 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 4.26425332E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 4.26425332E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.26425332E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 31, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7936251, 3.7936251, ]
- [ 3.7936251, 0.0000000, 3.7936251, ]
- [ 3.7936251, 3.7936251, 0.0000000, ]
lattice_lengths: [ 5.36500, 5.36500, 5.36500, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0919260E+02
convergence: {deltae: -6.214E-10, res2: 5.340E-09, residm: 5.184E-11, diffor: null, }
etotal : -2.35817298E+00
entropy : 0.00000000E+00
fermie : 2.84159263E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 4.26425332E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 4.26425332E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 4.26425332E-05, ]
pressure_GPa: -1.2546E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92736385
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.58725013896379E+00 7.58725013896379E+00 7.58725013896379E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.79362506948190E+00 3.79362506948190E+00
3.79362506948190E+00 0.00000000000000E+00 3.79362506948190E+00
3.79362506948190E+00 3.79362506948190E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09192602087812E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.36499602381987E+00 5.36499602381987E+00 5.36499602381987E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
4.26425331503709E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 4.26425331503709E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 4.26425331503706E-05
Total energy (etotal) [Ha]= -2.35817297838868E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.72067E-05
Relative =-1.15373E-05
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 31, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3582090799665 -2.358E+00 1.927E-03 3.403E-04
ETOT 2 -2.3582091614137 -8.145E-08 8.653E-10 1.468E-05
ETOT 3 -2.3582091672273 -5.814E-09 4.923E-10 5.111E-08
At SCF step 3, etot is converged :
for the second time, diff in etot= 5.814E-09 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.28082202E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.28082202E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.28082202E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 31, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7735781, 3.7735781, ]
- [ 3.7735781, 0.0000000, 3.7735781, ]
- [ 3.7735781, 3.7735781, 0.0000000, ]
lattice_lengths: [ 5.33665, 5.33665, 5.33665, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0747068E+02
convergence: {deltae: -5.814E-09, res2: 5.111E-08, residm: 4.923E-10, diffor: null, }
etotal : -2.35820917E+00
entropy : 0.00000000E+00
fermie : 2.90105483E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.28082202E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.28082202E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.28082202E-06, ]
pressure_GPa: 3.7683E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.91920490
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.54715614770044E+00 7.54715614770044E+00 7.54715614770044E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77357807385022E+00 3.77357807385022E+00
3.77357807385022E+00 0.00000000000000E+00 3.77357807385022E+00
3.77357807385022E+00 3.77357807385022E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07470684521666E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.33664529071272E+00 5.33664529071272E+00 5.33664529071272E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.28082202323682E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.28082202323682E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.28082202323677E-06
Total energy (etotal) [Ha]= -2.35820916722727E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-3.61888E-05
Relative =-1.53460E-05
--- Iteration: ( 4/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 31, itime: 4, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3582092000363 -2.358E+00 2.306E-09 3.161E-07
ETOT 2 -2.3582092001136 -7.730E-11 2.168E-13 1.381E-08
ETOT 3 -2.3582092001191 -5.500E-12 4.897E-13 4.554E-11
At SCF step 3, etot is converged :
for the second time, diff in etot= 5.500E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.45307548E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.45307548E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.45307548E-08 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 31, itime: 4, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7741537, 3.7741537, ]
- [ 3.7741537, 0.0000000, 3.7741537, ]
- [ 3.7741537, 3.7741537, 0.0000000, ]
lattice_lengths: [ 5.33746, 5.33746, 5.33746, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0751987E+02
convergence: {deltae: -5.500E-12, res2: 4.554E-11, residm: 4.897E-13, diffor: null, }
etotal : -2.35820920E+00
entropy : 0.00000000E+00
fermie : 2.89934352E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.45307548E-08, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.45307548E-08, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.45307548E-08, ]
pressure_GPa: 4.2751E-04
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.91944393
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.54830739632641E+00 7.54830739632641E+00 7.54830739632641E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77415369816320E+00 3.77415369816320E+00
3.77415369816320E+00 0.00000000000000E+00 3.77415369816320E+00
3.77415369816320E+00 3.77415369816320E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07519872990647E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.33745934642297E+00 5.33745934642297E+00 5.33745934642297E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.45307548347256E-08 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.45307548347527E-08 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.45307548346984E-08
Total energy (etotal) [Ha]= -2.35820920011913E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-3.28919E-08
Relative =-1.39478E-08
At Broyd/MD step 4, gradients are converged :
max grad (force/stress) = 1.4531E-06 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 87.110E-15; max= 48.974E-14
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.548307396326 7.548307396326 7.548307396326 bohr
= 3.994392252317 3.994392252317 3.994392252317 angstroms
prteigrs : about to open file tbase4_3o_DS31_EIG
Fermi (or HOMO) energy (hartree) = 0.28993 Average Vxc (hartree)= -0.36979
Eigenvalues (hartree) for nkpt= 28 k points:
kpt# 1, nband= 3, wtk= 0.02778, kpt= -0.0833 -0.1667 0.0000 (reduced coord)
-0.09900 0.59009 0.67177
occupation numbers for kpt# 1
2.00000 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 31, itime: 4, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.70489439045283E-01
hartree : 4.02781008485225E-03
xc : -1.12057096695142E+00
Ewald energy : -2.73331201953345E+00
psp_core : 1.60265792071268E-02
local_psp : 1.80577876674481E-01
non_local_psp : 4.24502054390921E-01
internal : -2.35825922708220E+00
'-kT*entropy' : 5.00269630706540E-05
total_energy : -2.35820920011913E+00
total_energy_eV : -6.41701413932676E+01
band_energy : 3.82094534513038E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.45307548E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.45307548E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.45307548E-08 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 4.2751E-04 GPa]
- sigma(1 1)= -4.27509567E-04 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -4.27509567E-04 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -4.27509567E-04 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 32 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 32, }
dimensions: {natom: 1, nkpt: 28, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 94, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 2.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 31.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_3o_DS31_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.361 89.328
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 32, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581162911358 -2.358E+00 2.663E-07 1.524E-03
ETOT 2 -2.3581167897798 -4.986E-07 1.303E-10 7.378E-05
ETOT 3 -2.3581168251550 -3.538E-08 3.855E-09 1.901E-07
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.538E-08 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 5.56052325E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 5.56052325E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 5.56052325E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 32, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -3.538E-08, res2: 1.901E-07, residm: 3.855E-09, diffor: null, }
etotal : -2.35811683E+00
entropy : 0.00000000E+00
fermie : 2.83348266E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 5.56052325E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 5.56052325E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 5.56052325E-05, ]
pressure_GPa: -1.6360E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92995954
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
5.56052324839385E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 5.56052324839385E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 5.56052324839384E-05
Total energy (etotal) [Ha]= -2.35811682515499E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 32, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581437612360 -2.358E+00 1.975E-08 3.549E-05
ETOT 2 -2.3581437699842 -8.748E-09 3.710E-11 1.550E-06
ETOT 3 -2.3581437706181 -6.338E-10 5.397E-11 5.406E-09
At SCF step 3, etot is converged :
for the second time, diff in etot= 6.338E-10 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 4.25227294E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 4.25227294E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.25227294E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 32, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7936610, 3.7936610, ]
- [ 3.7936610, 0.0000000, 3.7936610, ]
- [ 3.7936610, 3.7936610, 0.0000000, ]
lattice_lengths: [ 5.36505, 5.36505, 5.36505, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0919571E+02
convergence: {deltae: -6.338E-10, res2: 5.406E-09, residm: 5.397E-11, diffor: null, }
etotal : -2.35814377E+00
entropy : 0.00000000E+00
fermie : 2.85215603E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 4.25227294E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 4.25227294E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 4.25227294E-05, ]
pressure_GPa: -1.2511E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92732964
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.58732200699366E+00 7.58732200699366E+00 7.58732200699366E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.79366100349683E+00 3.79366100349683E+00
3.79366100349683E+00 0.00000000000000E+00 3.79366100349683E+00
3.79366100349683E+00 3.79366100349683E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09195705003115E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.36504684219114E+00 5.36504684219114E+00 5.36504684219114E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
4.25227294348012E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 4.25227294348012E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 4.25227294348012E-05
Total energy (etotal) [Ha]= -2.35814377061807E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.69455E-05
Relative =-1.14266E-05
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 32, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581798831113 -2.358E+00 1.943E-03 3.507E-04
ETOT 2 -2.3581799669200 -8.381E-08 8.957E-10 1.516E-05
ETOT 3 -2.3581799729389 -6.019E-09 5.185E-10 5.262E-08
At SCF step 3, etot is converged :
for the second time, diff in etot= 6.019E-09 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.39762396E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.39762396E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.39762396E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 32, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7734876, 3.7734876, ]
- [ 3.7734876, 0.0000000, 3.7734876, ]
- [ 3.7734876, 3.7734876, 0.0000000, ]
lattice_lengths: [ 5.33652, 5.33652, 5.33652, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0746295E+02
convergence: {deltae: -6.019E-09, res2: 5.262E-08, residm: 5.185E-10, diffor: null, }
etotal : -2.35817997E+00
entropy : 0.00000000E+00
fermie : 2.91222521E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.39762396E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.39762396E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.39762396E-06, ]
pressure_GPa: 4.1120E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.91907285
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.54697515662237E+00 7.54697515662237E+00 7.54697515662237E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77348757831118E+00 3.77348757831118E+00
3.77348757831118E+00 0.00000000000000E+00 3.77348757831118E+00
3.77348757831118E+00 3.77348757831118E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07462952827154E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.33651731069408E+00 5.33651731069408E+00 5.33651731069408E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.39762395652600E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.39762395652603E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.39762395652597E-06
Total energy (etotal) [Ha]= -2.35817997293893E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-3.62023E-05
Relative =-1.53519E-05
--- Iteration: ( 4/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 32, itime: 4, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581800121058 -2.358E+00 3.136E-09 3.858E-07
ETOT 2 -2.3581800122002 -9.440E-11 2.657E-13 1.687E-08
ETOT 3 -2.3581800122070 -6.763E-12 6.080E-13 5.548E-11
At SCF step 3, etot is converged :
for the second time, diff in etot= 6.763E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.37793194E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.37793194E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.37793194E-08 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 32, itime: 4, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7741197, 3.7741197, ]
- [ 3.7741197, 0.0000000, 3.7741197, ]
- [ 3.7741197, 3.7741197, 0.0000000, ]
lattice_lengths: [ 5.33741, 5.33741, 5.33741, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0751696E+02
convergence: {deltae: -6.763E-12, res2: 5.548E-11, residm: 6.080E-13, diffor: null, }
etotal : -2.35818001E+00
entropy : 0.00000000E+00
fermie : 2.91033705E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.37793194E-08, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.37793194E-08, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.37793194E-08, ]
pressure_GPa: 4.0540E-04
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.91933624
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.54823933022401E+00 7.54823933022401E+00 7.54823933022401E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77411966511200E+00 3.77411966511200E+00
3.77411966511200E+00 0.00000000000000E+00 3.77411966511200E+00
3.77411966511200E+00 3.77411966511200E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07516964367969E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.33741121642040E+00 5.33741121642040E+00 5.33741121642040E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.37793194221068E-08 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.37793194220526E-08 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.37793194220255E-08
Total energy (etotal) [Ha]= -2.35818001220699E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-3.92681E-08
Relative =-1.66519E-08
At Broyd/MD step 4, gradients are converged :
max grad (force/stress) = 1.3779E-06 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 10.751E-14; max= 60.796E-14
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.548239330224 7.548239330224 7.548239330224 bohr
= 3.994356233286 3.994356233286 3.994356233286 angstroms
prteigrs : about to open file tbase4_3o_DS32_EIG
Fermi (or HOMO) energy (hartree) = 0.29103 Average Vxc (hartree)= -0.36980
Eigenvalues (hartree) for nkpt= 28 k points:
kpt# 1, nband= 3, wtk= 0.02778, kpt= -0.0833 -0.1667 0.0000 (reduced coord)
-0.09901 0.59009 0.67178
occupation numbers for kpt# 1
2.00000 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 32, itime: 4, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.70405167011029E-01
hartree : 3.98807685208577E-03
xc : -1.12054240168018E+00
Ewald energy : -2.73333666712179E+00
psp_core : 1.60270127691420E-02
local_psp : 1.80727733947943E-01
non_local_psp : 4.24487404991157E-01
internal : -2.35824367323061E+00
'-kT*entropy' : 6.36610236194626E-05
total_energy : -2.35818001220699E+00
total_energy_eV : -6.41693471497167E+01
band_energy : 3.82101678455056E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.37793194E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.37793194E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.37793194E-08 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 4.0540E-04 GPa]
- sigma(1 1)= -4.05401574E-04 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -4.05401574E-04 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -4.05401574E-04 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 33 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 33, }
dimensions: {natom: 1, nkpt: 28, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 94, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 3.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 32.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_3o_DS32_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.361 89.328
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 33, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581322270479 -2.358E+00 2.654E-07 1.524E-03
ETOT 2 -2.3581327248372 -4.978E-07 1.292E-10 7.372E-05
ETOT 3 -2.3581327601624 -3.533E-08 3.845E-09 1.908E-07
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.533E-08 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 5.35462655E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 5.35462655E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 5.35462655E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 33, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -3.533E-08, res2: 1.908E-07, residm: 3.845E-09, diffor: null, }
etotal : -2.35813276E+00
entropy : 0.00000000E+00
fermie : 2.84769590E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 5.35462655E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 5.35462655E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 5.35462655E-05, ]
pressure_GPa: -1.5754E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93004443
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
5.35462655267304E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 5.35462655267304E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 5.35462655267304E-05
Total energy (etotal) [Ha]= -2.35813276016238E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 33, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581577594428 -2.358E+00 1.841E-08 3.317E-05
ETOT 2 -2.3581577676275 -8.185E-09 3.243E-11 1.448E-06
ETOT 3 -2.3581577682210 -5.935E-10 5.057E-11 5.055E-09
At SCF step 3, etot is converged :
for the second time, diff in etot= 5.935E-10 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 4.10263641E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 4.10263641E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.10263641E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 33, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7938957, 3.7938957, ]
- [ 3.7938957, 0.0000000, 3.7938957, ]
- [ 3.7938957, 3.7938957, 0.0000000, ]
lattice_lengths: [ 5.36538, 5.36538, 5.36538, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0921597E+02
convergence: {deltae: -5.935E-10, res2: 5.055E-09, residm: 5.057E-11, diffor: null, }
etotal : -2.35815777E+00
entropy : 0.00000000E+00
fermie : 2.86567358E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 4.10263641E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 4.10263641E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 4.10263641E-05, ]
pressure_GPa: -1.2070E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92750400
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.58779145145991E+00 7.58779145145991E+00 7.58779145145991E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.79389572572995E+00 3.79389572572995E+00
3.79389572572995E+00 0.00000000000000E+00 3.79389572572995E+00
3.79389572572995E+00 3.79389572572995E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09215974799674E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.36537878955661E+00 5.36537878955661E+00 5.36537878955661E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
4.10263640812270E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 4.10263640812270E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 4.10263640812270E-05
Total energy (etotal) [Ha]= -2.35815776822101E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.50081E-05
Relative =-1.06050E-05
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 33, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581916466336 -2.358E+00 5.427E-07 3.335E-04
ETOT 2 -2.3581917264944 -7.986E-08 7.338E-10 1.442E-05
ETOT 3 -2.3581917322357 -5.741E-09 4.945E-10 5.010E-08
At SCF step 3, etot is converged :
for the second time, diff in etot= 5.741E-09 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.29630683E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.29630683E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.29630683E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 33, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7742979, 3.7742979, ]
- [ 3.7742979, 0.0000000, 3.7742979, ]
- [ 3.7742979, 3.7742979, 0.0000000, ]
lattice_lengths: [ 5.33766, 5.33766, 5.33766, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0753220E+02
convergence: {deltae: -5.741E-09, res2: 5.010E-08, residm: 4.945E-10, diffor: null, }
etotal : -2.35819173E+00
entropy : 0.00000000E+00
fermie : 2.92403583E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.29630683E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.29630683E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.29630683E-06, ]
pressure_GPa: 3.8139E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.91945637
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.54859582245447E+00 7.54859582245447E+00 7.54859582245447E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77429791122724E+00 3.77429791122724E+00
3.77429791122724E+00 0.00000000000000E+00 3.77429791122724E+00
3.77429791122724E+00 3.77429791122724E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07532198691237E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.33766329449400E+00 5.33766329449400E+00 5.33766329449400E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.29630683085908E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.29630683085914E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.29630683085908E-06
Total energy (etotal) [Ha]= -2.35819173223572E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-3.39640E-05
Relative =-1.44027E-05
--- Iteration: ( 4/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 33, itime: 4, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3581917662250 -2.358E+00 1.600E-09 3.408E-07
ETOT 2 -2.3581917663084 -8.347E-11 2.169E-13 1.490E-08
ETOT 3 -2.3581917663144 -5.989E-12 5.383E-13 4.912E-11
At SCF step 3, etot is converged :
for the second time, diff in etot= 5.989E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.03514352E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.03514352E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.03514352E-08 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 33, itime: 4, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7748892, 3.7748892, ]
- [ 3.7748892, 0.0000000, 3.7748892, ]
- [ 3.7748892, 3.7748892, 0.0000000, ]
lattice_lengths: [ 5.33850, 5.33850, 5.33850, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0758275E+02
convergence: {deltae: -5.989E-12, res2: 4.912E-11, residm: 5.383E-13, diffor: null, }
etotal : -2.35819177E+00
entropy : 0.00000000E+00
fermie : 2.92226900E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.03514352E-08, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.03514352E-08, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.03514352E-08, ]
pressure_GPa: 3.0455E-04
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.91970387
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.54977840064516E+00 7.54977840064516E+00 7.54977840064516E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77488920032258E+00 3.77488920032258E+00
3.77488920032258E+00 0.00000000000000E+00 3.77488920032258E+00
3.77488920032258E+00 3.77488920032258E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07582745240143E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.33849950355192E+00 5.33849950355192E+00 5.33849950355192E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.03514352191910E-08 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.03514352191639E-08 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.03514352191639E-08
Total energy (etotal) [Ha]= -2.35819176631443E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-3.40787E-08
Relative =-1.44512E-08
At Broyd/MD step 4, gradients are converged :
max grad (force/stress) = 1.0351E-06 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 95.376E-15; max= 53.830E-14
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.549778400645 7.549778400645 7.549778400645 bohr
= 3.995170674279 3.995170674279 3.995170674279 angstroms
prteigrs : about to open file tbase4_3o_DS33_EIG
Fermi (or HOMO) energy (hartree) = 0.29223 Average Vxc (hartree)= -0.36973
Eigenvalues (hartree) for nkpt= 28 k points:
kpt# 1, nband= 3, wtk= 0.02778, kpt= -0.0833 -0.1667 0.0000 (reduced coord)
-0.09909 0.58975 0.67139
occupation numbers for kpt# 1
2.00000 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 33, itime: 4, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.70305331372897E-01
hartree : 3.97389070474900E-03
xc : -1.12037352614864E+00
Ewald energy : -2.73277945903010E+00
psp_core : 1.60172131411816E-02
local_psp : 1.80399244544630E-01
non_local_psp : 4.24451644042626E-01
internal : -2.35800566137265E+00
'-kT*entropy' : -1.86104941773870E-04
total_energy : -2.35819176631443E+00
total_energy_eV : -6.41696669952741E+01
band_energy : 3.81822790191797E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.03514352E-08 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.03514352E-08 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.03514352E-08 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 3.0455E-04 GPa]
- sigma(1 1)= -3.04549739E-04 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -3.04549739E-04 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -3.04549739E-04 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 34 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 34, }
dimensions: {natom: 1, nkpt: 28, mband: 3, nsppol: 1, nspinor: 1, nspden: 1, mpw: 94, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 3.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 4.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 1, iscf: 7, paral_kgb: 0, }
...
mkfilename : getwfk/=0, take file _WFK from output of DATASET 33.
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 3.8000000 3.8000000 G(1)= -0.1315789 0.1315789 0.1315789
R(2)= 3.8000000 0.0000000 3.8000000 G(2)= 0.1315789 -0.1315789 0.1315789
R(3)= 3.8000000 3.8000000 0.0000000 G(3)= 0.1315789 0.1315789 -0.1315789
Unit cell volume ucvol= 1.0974400E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 15 15 15
ecut(hartree)= 6.615 => boxcut(ratio)= 2.26154
getcut : COMMENT -
Note that boxcut > 2.2 ; recall that boxcut=Gcut(box)/Gcut(sphere) = 2
is sufficient for exact treatment of convolution.
Such a large boxcut is a waste : you could raise ecut
e.g. ecut= 8.458196 Hartrees makes boxcut=2
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file tbase4_3o_DS33_WFK
_setup2: Arith. and geom. avg. npw (full set) are 89.361 89.328
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 34, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3582345524438 -2.358E+00 2.504E-07 1.433E-03
ETOT 2 -2.3582350197186 -4.673E-07 1.217E-10 6.932E-05
ETOT 3 -2.3582350528981 -3.318E-08 3.615E-09 1.798E-07
At SCF step 3, etot is converged :
for the second time, diff in etot= 3.318E-08 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 5.08515194E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 5.08515194E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 5.08515194E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 34, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.8000000, 3.8000000, ]
- [ 3.8000000, 0.0000000, 3.8000000, ]
- [ 3.8000000, 3.8000000, 0.0000000, ]
lattice_lengths: [ 5.37401, 5.37401, 5.37401, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0974400E+02
convergence: {deltae: -3.318E-08, res2: 1.798E-07, residm: 3.615E-09, diffor: null, }
etotal : -2.35823505E+00
entropy : 0.00000000E+00
fermie : 2.85806090E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 5.08515194E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 5.08515194E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 5.08515194E-05, ]
pressure_GPa: -1.4961E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.93023973
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.60000000000000E+00 7.60000000000000E+00 7.60000000000000E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.80000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 0.00000000000000E+00 3.80000000000000E+00
3.80000000000000E+00 3.80000000000000E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09744000000000E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.37401153701776E+00 5.37401153701776E+00 5.37401153701776E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
5.08515194433603E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 5.08515194433603E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 5.08515194433603E-05
Total energy (etotal) [Ha]= -2.35823505289806E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 34, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3582576047498 -2.358E+00 1.677E-08 2.991E-05
ETOT 2 -2.3582576121270 -7.377E-09 2.746E-11 1.306E-06
ETOT 3 -2.3582576126624 -5.354E-10 4.564E-11 4.567E-09
At SCF step 3, etot is converged :
for the second time, diff in etot= 5.354E-10 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 3.89808487E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 3.89808487E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.89808487E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 34, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7942029, 3.7942029, ]
- [ 3.7942029, 0.0000000, 3.7942029, ]
- [ 3.7942029, 3.7942029, 0.0000000, ]
lattice_lengths: [ 5.36581, 5.36581, 5.36581, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0924251E+02
convergence: {deltae: -5.354E-10, res2: 4.567E-09, residm: 4.564E-11, diffor: null, }
etotal : -2.35825761E+00
entropy : 0.00000000E+00
fermie : 2.87518866E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 3.89808487E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 3.89808487E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.89808487E-05, ]
pressure_GPa: -1.1469E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92782583
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.58840585356691E+00 7.58840585356691E+00 7.58840585356691E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.79420292678346E+00 3.79420292678346E+00
3.79420292678346E+00 0.00000000000000E+00 3.79420292678346E+00
3.79420292678346E+00 3.79420292678346E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.09242507405005E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.36581323745286E+00 5.36581323745286E+00 5.36581323745286E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
3.89808486796162E-05 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.89808486796161E-05 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 3.89808486796162E-05
Total energy (etotal) [Ha]= -2.35825761266239E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.25598E-05
Relative =-9.56633E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 34, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3582883052245 -2.358E+00 2.046E-07 3.028E-04
ETOT 2 -2.3582883777965 -7.257E-08 5.463E-10 1.309E-05
ETOT 3 -2.3582883830198 -5.223E-09 4.496E-10 4.560E-08
At SCF step 3, etot is converged :
for the second time, diff in etot= 5.223E-09 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -1.16342938E-06 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -1.16342938E-06 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -1.16342938E-06 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 34, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7755337, 3.7755337, ]
- [ 3.7755337, 0.0000000, 3.7755337, ]
- [ 3.7755337, 3.7755337, 0.0000000, ]
lattice_lengths: [ 5.33941, 5.33941, 5.33941, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0763786E+02
convergence: {deltae: -5.223E-09, res2: 4.560E-08, residm: 4.496E-10, diffor: null, }
etotal : -2.35828838E+00
entropy : 0.00000000E+00
fermie : 2.93094889E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -1.16342938E-06, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -1.16342938E-06, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -1.16342938E-06, ]
pressure_GPa: 3.4229E-02
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92015190
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.55106732900262E+00 7.55106732900262E+00 7.55106732900262E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77553366450131E+00 3.77553366450131E+00
3.77553366450131E+00 0.00000000000000E+00 3.77553366450131E+00
3.77553366450131E+00 3.77553366450131E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07637855517078E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.33941091353394E+00 5.33941091353394E+00 5.33941091353394E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-1.16342937959356E-06 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -1.16342937959353E-06 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -1.16342937959347E-06
Total energy (etotal) [Ha]= -2.35828838301981E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-3.07704E-05
Relative =-1.30478E-05
--- Iteration: ( 4/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 34, itime: 4, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldfe: 1.00E-06, }
...
iter Etot(hartree) deltaE(h) residm vres2
ETOT 1 -2.3582884105168 -2.358E+00 6.739E-10 2.780E-07
ETOT 2 -2.3582884105849 -6.808E-11 1.657E-13 1.216E-08
ETOT 3 -2.3582884105898 -4.889E-12 4.398E-13 4.023E-11
At SCF step 3, etot is converged :
for the second time, diff in etot= 4.889E-12 < toldfe= 1.000E-06
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -5.11878000E-09 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -5.11878000E-09 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -5.11878000E-09 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 34, itime: 4, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 0.0000000, 3.7760670, 3.7760670, ]
- [ 3.7760670, 0.0000000, 3.7760670, ]
- [ 3.7760670, 3.7760670, 0.0000000, ]
lattice_lengths: [ 5.34017, 5.34017, 5.34017, ]
lattice_angles: [ 60.000, 60.000, 60.000, ] # degrees, (23, 13, 12)
lattice_volume: 1.0768348E+02
convergence: {deltae: -4.889E-12, res2: 4.023E-11, residm: 4.398E-13, diffor: null, }
etotal : -2.35828841E+00
entropy : 0.00000000E+00
fermie : 2.92935110E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ -5.11878000E-09, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, -5.11878000E-09, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -5.11878000E-09, ]
pressure_GPa: 1.5060E-04
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
force_length_stats: {min: 0.00000000E+00, max: 0.00000000E+00, mean: 0.00000000E+00, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.92037578
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Cartesian forces (fcart) [Ha/bohr]; max,rms= 0.00000E+00 0.00000E+00 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
Gradient of E wrt nuclear positions in reduced coordinates (gred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
Scale of Primitive Cell (acell) [bohr]
7.55213400328296E+00 7.55213400328296E+00 7.55213400328296E+00
Real space primitive translations (rprimd) [bohr]
0.00000000000000E+00 3.77606700164148E+00 3.77606700164148E+00
3.77606700164148E+00 0.00000000000000E+00 3.77606700164148E+00
3.77606700164148E+00 3.77606700164148E+00 0.00000000000000E+00
Unitary Cell Volume (ucvol) [Bohr^3]= 1.07683477180887E+02
Angles (23,13,12)= [degrees]
6.00000000000000E+01 6.00000000000000E+01 6.00000000000000E+01
Lengths [Bohr]
5.34016516615089E+00 5.34016516615089E+00 5.34016516615089E+00
Stress tensor in cartesian coordinates (strten) [Ha/bohr^3]
-5.11877999869229E-09 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 -5.11877999869229E-09 0.00000000000000E+00
0.00000000000000E+00 0.00000000000000E+00 -5.11877999869229E-09
Total energy (etotal) [Ha]= -2.35828841058976E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.75699E-08
Relative =-1.16907E-08
At Broyd/MD step 4, gradients are converged :
max grad (force/stress) = 5.1188E-07 < tolmxf= 5.0000E-05 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 77.935E-15; max= 43.975E-14
reduced coordinates (array xred) for 1 atoms
0.000000000000 0.000000000000 0.000000000000
rms dE/dt= 0.0000E+00; max dE/dt= 0.0000E+00; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
frms,max,avg= 0.0000000E+00 0.0000000E+00 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.552134003283 7.552134003283 7.552134003283 bohr
= 3.996417205512 3.996417205512 3.996417205512 angstroms
prteigrs : about to open file tbase4_3o_DS34_EIG
Fermi (or HOMO) energy (hartree) = 0.29294 Average Vxc (hartree)= -0.36961
Eigenvalues (hartree) for nkpt= 28 k points:
kpt# 1, nband= 3, wtk= 0.02778, kpt= -0.0833 -0.1667 0.0000 (reduced coord)
-0.09922 0.58923 0.67080
occupation numbers for kpt# 1
2.00000 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 34, itime: 4, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.70006049689987E-01
hartree : 3.95970887284377E-03
xc : -1.12012851878488E+00
Ewald energy : -2.73192707181089E+00
psp_core : 1.60022299236327E-02
local_psp : 1.79793783822399E-01
non_local_psp : 4.24498139546681E-01
internal : -2.35779567874022E+00
'-kT*entropy' : -4.92731849536055E-04
total_energy : -2.35828841058976E+00
total_energy_eV : -6.41722968199786E+01
band_energy : 3.81250156382240E-01
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= -5.11878000E-09 sigma(3 2)= 0.00000000E+00
sigma(2 2)= -5.11878000E-09 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -5.11878000E-09 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= 1.5060E-04 GPa]
- sigma(1 1)= -1.50599707E-04 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= -1.50599707E-04 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= -1.50599707E-04 sigma(2 1)= 0.00000000E+00
== END DATASET(S) ==============================================================
================================================================================
-outvars: echo values of variables after computation --------
acell11 7.6022357792E+00 7.6022357792E+00 7.6022357792E+00 Bohr
acell12 7.6022341271E+00 7.6022341271E+00 7.6022341271E+00 Bohr
acell13 7.6022341214E+00 7.6022341214E+00 7.6022341214E+00 Bohr
acell14 7.6022357148E+00 7.6022357148E+00 7.6022357148E+00 Bohr
acell21 7.5604102145E+00 7.5604102145E+00 7.5604102145E+00 Bohr
acell22 7.5605496029E+00 7.5605496029E+00 7.5605496029E+00 Bohr
acell23 7.5565044147E+00 7.5565044147E+00 7.5565044147E+00 Bohr
acell24 7.5593333886E+00 7.5593333886E+00 7.5593333886E+00 Bohr
acell31 7.5483073963E+00 7.5483073963E+00 7.5483073963E+00 Bohr
acell32 7.5482393302E+00 7.5482393302E+00 7.5482393302E+00 Bohr
acell33 7.5497784006E+00 7.5497784006E+00 7.5497784006E+00 Bohr
acell34 7.5521340033E+00 7.5521340033E+00 7.5521340033E+00 Bohr
amu 2.69815390E+01
dilatmx 1.05000000E+00
ecut 6.00000000E+00 Hartree
ecutsm 5.00000000E-01 Hartree
etotal11 -2.3516656074E+00
etotal12 -2.3532597160E+00
etotal13 -2.3548538247E+00
etotal14 -2.3564479440E+00
etotal21 -2.3568282638E+00
etotal22 -2.3574128355E+00
etotal23 -2.3576771874E+00
etotal24 -2.3578584768E+00
etotal31 -2.3582092001E+00
etotal32 -2.3581800122E+00
etotal33 -2.3581917663E+00
etotal34 -2.3582884106E+00
fcart11 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart12 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart13 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart14 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart21 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart22 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart23 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart24 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart31 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart32 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart33 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
fcart34 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
- fftalg 512
getwfk -1
ionmov 2
ixc -1012
jdtset 11 12 13 14 21 22 23 24 31 32
33 34
kpt11 -2.50000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
kpt12 -2.50000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
kpt13 -2.50000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
kpt14 -2.50000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
kpt21 -1.25000000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.75000000E-01 0.00000000E+00
-3.75000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 1.25000000E-01
-1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
kpt22 -1.25000000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.75000000E-01 0.00000000E+00
-3.75000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 1.25000000E-01
-1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
kpt23 -1.25000000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.75000000E-01 0.00000000E+00
-3.75000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 1.25000000E-01
-1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
kpt24 -1.25000000E-01 -2.50000000E-01 0.00000000E+00
-1.25000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 -3.75000000E-01 0.00000000E+00
-1.25000000E-01 -3.75000000E-01 1.25000000E-01
-1.25000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.75000000E-01 0.00000000E+00
-3.75000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 1.25000000E-01
-1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
kpt31 -8.33333333E-02 -1.66666667E-01 0.00000000E+00
-8.33333333E-02 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -2.50000000E-01 0.00000000E+00
-8.33333333E-02 -2.50000000E-01 8.33333333E-02
-8.33333333E-02 5.00000000E-01 0.00000000E+00
-1.66666667E-01 -4.16666667E-01 0.00000000E+00
-8.33333333E-02 -4.16666667E-01 8.33333333E-02
-2.50000000E-01 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -3.33333333E-01 8.33333333E-02
-8.33333333E-02 -3.33333333E-01 1.66666667E-01
-8.33333333E-02 3.33333333E-01 0.00000000E+00
-1.66666667E-01 4.16666667E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 0.00000000E+00
-1.66666667E-01 5.00000000E-01 8.33333333E-02
-3.33333333E-01 -4.16666667E-01 0.00000000E+00
-2.50000000E-01 -4.16666667E-01 8.33333333E-02
-1.66666667E-01 -4.16666667E-01 1.66666667E-01
-8.33333333E-02 -4.16666667E-01 2.50000000E-01
-8.33333333E-02 1.66666667E-01 0.00000000E+00
-1.66666667E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.33333333E-01 0.00000000E+00
-3.33333333E-01 4.16666667E-01 0.00000000E+00
-4.16666667E-01 5.00000000E-01 0.00000000E+00
-3.33333333E-01 5.00000000E-01 8.33333333E-02
-2.50000000E-01 5.00000000E-01 1.66666667E-01
-8.33333333E-02 0.00000000E+00 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
-4.16666667E-01 0.00000000E+00 0.00000000E+00
kpt32 -8.33333333E-02 -1.66666667E-01 0.00000000E+00
-8.33333333E-02 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -2.50000000E-01 0.00000000E+00
-8.33333333E-02 -2.50000000E-01 8.33333333E-02
-8.33333333E-02 5.00000000E-01 0.00000000E+00
-1.66666667E-01 -4.16666667E-01 0.00000000E+00
-8.33333333E-02 -4.16666667E-01 8.33333333E-02
-2.50000000E-01 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -3.33333333E-01 8.33333333E-02
-8.33333333E-02 -3.33333333E-01 1.66666667E-01
-8.33333333E-02 3.33333333E-01 0.00000000E+00
-1.66666667E-01 4.16666667E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 0.00000000E+00
-1.66666667E-01 5.00000000E-01 8.33333333E-02
-3.33333333E-01 -4.16666667E-01 0.00000000E+00
-2.50000000E-01 -4.16666667E-01 8.33333333E-02
-1.66666667E-01 -4.16666667E-01 1.66666667E-01
-8.33333333E-02 -4.16666667E-01 2.50000000E-01
-8.33333333E-02 1.66666667E-01 0.00000000E+00
-1.66666667E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.33333333E-01 0.00000000E+00
-3.33333333E-01 4.16666667E-01 0.00000000E+00
-4.16666667E-01 5.00000000E-01 0.00000000E+00
-3.33333333E-01 5.00000000E-01 8.33333333E-02
-2.50000000E-01 5.00000000E-01 1.66666667E-01
-8.33333333E-02 0.00000000E+00 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
-4.16666667E-01 0.00000000E+00 0.00000000E+00
kpt33 -8.33333333E-02 -1.66666667E-01 0.00000000E+00
-8.33333333E-02 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -2.50000000E-01 0.00000000E+00
-8.33333333E-02 -2.50000000E-01 8.33333333E-02
-8.33333333E-02 5.00000000E-01 0.00000000E+00
-1.66666667E-01 -4.16666667E-01 0.00000000E+00
-8.33333333E-02 -4.16666667E-01 8.33333333E-02
-2.50000000E-01 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -3.33333333E-01 8.33333333E-02
-8.33333333E-02 -3.33333333E-01 1.66666667E-01
-8.33333333E-02 3.33333333E-01 0.00000000E+00
-1.66666667E-01 4.16666667E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 0.00000000E+00
-1.66666667E-01 5.00000000E-01 8.33333333E-02
-3.33333333E-01 -4.16666667E-01 0.00000000E+00
-2.50000000E-01 -4.16666667E-01 8.33333333E-02
-1.66666667E-01 -4.16666667E-01 1.66666667E-01
-8.33333333E-02 -4.16666667E-01 2.50000000E-01
-8.33333333E-02 1.66666667E-01 0.00000000E+00
-1.66666667E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.33333333E-01 0.00000000E+00
-3.33333333E-01 4.16666667E-01 0.00000000E+00
-4.16666667E-01 5.00000000E-01 0.00000000E+00
-3.33333333E-01 5.00000000E-01 8.33333333E-02
-2.50000000E-01 5.00000000E-01 1.66666667E-01
-8.33333333E-02 0.00000000E+00 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
-4.16666667E-01 0.00000000E+00 0.00000000E+00
kpt34 -8.33333333E-02 -1.66666667E-01 0.00000000E+00
-8.33333333E-02 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -2.50000000E-01 0.00000000E+00
-8.33333333E-02 -2.50000000E-01 8.33333333E-02
-8.33333333E-02 5.00000000E-01 0.00000000E+00
-1.66666667E-01 -4.16666667E-01 0.00000000E+00
-8.33333333E-02 -4.16666667E-01 8.33333333E-02
-2.50000000E-01 -3.33333333E-01 0.00000000E+00
-1.66666667E-01 -3.33333333E-01 8.33333333E-02
-8.33333333E-02 -3.33333333E-01 1.66666667E-01
-8.33333333E-02 3.33333333E-01 0.00000000E+00
-1.66666667E-01 4.16666667E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 0.00000000E+00
-1.66666667E-01 5.00000000E-01 8.33333333E-02
-3.33333333E-01 -4.16666667E-01 0.00000000E+00
-2.50000000E-01 -4.16666667E-01 8.33333333E-02
-1.66666667E-01 -4.16666667E-01 1.66666667E-01
-8.33333333E-02 -4.16666667E-01 2.50000000E-01
-8.33333333E-02 1.66666667E-01 0.00000000E+00
-1.66666667E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 3.33333333E-01 0.00000000E+00
-3.33333333E-01 4.16666667E-01 0.00000000E+00
-4.16666667E-01 5.00000000E-01 0.00000000E+00
-3.33333333E-01 5.00000000E-01 8.33333333E-02
-2.50000000E-01 5.00000000E-01 1.66666667E-01
-8.33333333E-02 0.00000000E+00 0.00000000E+00
-2.50000000E-01 0.00000000E+00 0.00000000E+00
-4.16666667E-01 0.00000000E+00 0.00000000E+00
kptrlatt11 2 -2 2 -2 2 2 -2 -2 2
kptrlatt12 2 -2 2 -2 2 2 -2 -2 2
kptrlatt13 2 -2 2 -2 2 2 -2 -2 2
kptrlatt14 2 -2 2 -2 2 2 -2 -2 2
kptrlatt21 4 -4 4 -4 4 4 -4 -4 4
kptrlatt22 4 -4 4 -4 4 4 -4 -4 4
kptrlatt23 4 -4 4 -4 4 4 -4 -4 4
kptrlatt24 4 -4 4 -4 4 4 -4 -4 4
kptrlatt31 6 -6 6 -6 6 6 -6 -6 6
kptrlatt32 6 -6 6 -6 6 6 -6 -6 6
kptrlatt33 6 -6 6 -6 6 6 -6 -6 6
kptrlatt34 6 -6 6 -6 6 6 -6 -6 6
kptrlen11 1.52000000E+01
kptrlen12 1.52000000E+01
kptrlen13 1.52000000E+01
kptrlen14 1.52000000E+01
kptrlen21 3.04000000E+01
kptrlen22 3.04000000E+01
kptrlen23 3.04000000E+01
kptrlen24 3.04000000E+01
kptrlen31 4.56000000E+01
kptrlen32 4.56000000E+01
kptrlen33 4.56000000E+01
kptrlen34 4.56000000E+01
P mkmem11 2
P mkmem12 2
P mkmem13 2
P mkmem14 2
P mkmem21 10
P mkmem22 10
P mkmem23 10
P mkmem24 10
P mkmem31 28
P mkmem32 28
P mkmem33 28
P mkmem34 28
natom 1
nband11 3
nband12 3
nband13 3
nband14 3
nband21 3
nband22 3
nband23 3
nband24 3
nband31 3
nband32 3
nband33 3
nband34 3
ndtset 12
ngfft 15 15 15
nkpt11 2
nkpt12 2
nkpt13 2
nkpt14 2
nkpt21 10
nkpt22 10
nkpt23 10
nkpt24 10
nkpt31 28
nkpt32 28
nkpt33 28
nkpt34 28
nstep 10
nsym 48
ntime 10
ntypat 1
occ11 2.000000 1.333333 0.000000
2.000000 0.000000 0.000000
occ12 2.000000 1.333333 0.000000
2.000000 0.000000 0.000000
occ13 2.000000 1.333333 0.000000
2.000000 0.000000 0.000000
occ14 2.000000 1.333331 0.000001
2.000000 0.000004 0.000000
occ21 2.000000 0.000000 0.000000
2.000000 2.000000 0.000000
2.000000 0.000000 0.000000
2.000000 0.981890 0.000000
2.000000 0.000000 0.000000
2.000000 2.000000 0.000000
2.000000 0.036218 0.000000
2.000000 2.000000 0.000000
2.000000 0.000000 0.000000
2.000000 2.000003 0.000000
occ22 2.000000 0.000000 0.000000
2.000000 2.000000 0.000000
2.000000 0.000000 0.000000
2.000000 0.836024 0.000000
2.000000 0.000000 0.000000
2.000000 2.000001 0.000000
2.000000 0.233131 0.000001
2.000000 2.037328 0.001255
2.000000 0.000000 0.000000
2.000000 2.052962 0.000000
occ23 2.000000 0.000000 0.000000
2.000000 2.000003 0.000000
2.000000 0.000000 0.000000
2.000000 0.683352 0.000000
2.000000 0.000492 0.000000
2.000000 2.004794 0.000086
2.000000 0.286993 0.000558
2.000002 2.141791 0.005253
2.000000 0.000000 0.000000
2.000000 2.138835 0.000000
occ24 2.000000 0.000000 0.000000
2.000000 2.001141 0.000000
2.000000 0.000159 0.000000
2.000000 0.699061 0.000000
2.000000 0.003949 0.000000
2.000002 2.049323 0.002027
2.000002 0.380848 0.004112
2.000824 2.072295 0.000816
2.000000 0.000000 0.000000
2.000000 2.037334 0.000000
occ31 2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 2.000000 0.000000
2.000000 0.002239 0.000000
2.000000 2.000000 0.000000
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 0.001222 0.000000
2.000000 1.838737 0.000000
2.000000 2.000000 0.000000
2.000000 2.006374 0.000000
2.000000 2.000000 0.000000
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 2.115954 0.000000
2.000000 2.000000 0.000000
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 2.001621 0.000000
2.000000 2.000000 0.002351
2.000000 2.115951 0.000000
2.000000 0.564572 0.000000
2.000000 2.001280 0.000001
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 2.000000 0.000000
occ32 2.000000 0.000000 0.000000
2.000000 0.000072 0.000000
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 2.000000 0.000000
2.000000 0.204110 0.000000
2.000000 2.000432 0.000000
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 0.000109 0.000000
2.000000 1.476903 0.000000
2.000000 2.000000 0.000000
2.000000 2.139484 0.000000
2.000000 2.000000 0.000000
2.000000 0.000000 0.000000
2.000000 0.000227 0.000000
2.000000 1.768225 0.000000
2.000000 2.000174 0.000322
2.000000 0.000000 0.000000
2.000000 0.000001 0.000000
2.000000 2.122615 0.000000
2.000000 2.018724 0.002443
2.000000 2.002480 0.000000
2.000000 0.786326 0.003299
2.000000 2.118976 0.005162
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 2.000002 0.000000
occ33 2.000000 0.000000 0.000000
2.000000 0.004083 0.000000
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 2.000000 0.000000
2.000000 0.421656 0.000000
2.000000 2.029264 0.000000
2.000000 0.000000 0.000000
2.000000 0.000100 0.000000
2.000000 0.059728 0.000000
2.000000 1.351705 0.000000
2.000000 2.000002 0.000000
2.000000 2.071980 0.000000
2.000000 2.000122 0.000000
2.000000 0.000015 0.000000
2.000000 0.004991 0.000000
2.000000 1.568117 0.000008
2.000002 2.020446 0.005172
2.000000 0.000000 0.000000
2.000000 0.001187 0.000000
2.000000 2.111993 0.000133
2.000053 2.111454 0.088112
2.000000 1.784546 0.000000
2.000001 0.884894 0.001621
2.001453 2.116578 0.000215
2.000000 0.000000 0.000000
2.000000 0.000000 0.000000
2.000000 2.002962 0.000000
occ34 2.000000 0.000000 0.000000
2.000000 0.003410 0.000000
2.000000 0.000000 0.000000
2.000000 0.000005 0.000000
2.000000 2.000067 0.000000
2.000000 0.551799 0.000000
2.000000 2.100586 0.000000
2.000000 0.000000 0.000000
2.000000 0.002413 0.000000
2.000000 0.172025 0.000007
2.000000 1.271224 0.000000
2.000000 2.000829 0.000000
2.000000 1.926538 0.000013
2.000003 2.006735 0.000000
2.000000 0.001053 0.000007
2.000000 0.001695 0.000158
2.000000 1.440111 0.000786
2.000707 2.086495 0.001104
2.000000 0.000000 0.000000
2.000000 0.005137 0.000000
2.000000 1.991280 0.002703
2.004316 2.139641 0.213636
2.000037 1.623027 0.000087
2.000499 0.919525 0.007129
2.024305 2.000195 0.028990
2.000000 0.000000 0.000000
2.000000 0.000041 0.000000
2.000000 2.034715 0.000000
occopt 4
optcell 1
rprim 0.0000000000E+00 5.0000000000E-01 5.0000000000E-01
5.0000000000E-01 0.0000000000E+00 5.0000000000E-01
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
shiftk 5.00000000E-01 5.00000000E-01 5.00000000E-01
spgroup 225
strten11 -2.9882263317E-10 -2.9882263317E-10 -2.9882263317E-10
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten12 -2.0817727147E-09 -2.0817727147E-09 -2.0817727147E-09
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten13 -2.0876928107E-09 -2.0876928107E-09 -2.0876928107E-09
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten14 -2.1027695493E-09 -2.1027695493E-09 -2.1027695494E-09
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten21 2.0964325538E-07 2.0964325538E-07 2.0964325538E-07
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten22 -3.0786189791E-08 -3.0786189791E-08 -3.0786189791E-08
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten23 -1.5530850695E-07 -1.5530850695E-07 -1.5530850695E-07
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten24 -6.5450192351E-08 -6.5450192351E-08 -6.5450192351E-08
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten31 -1.4530754835E-08 -1.4530754835E-08 -1.4530754835E-08
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten32 -1.3779319422E-08 -1.3779319422E-08 -1.3779319422E-08
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten33 -1.0351435219E-08 -1.0351435219E-08 -1.0351435219E-08
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten34 -5.1187799987E-09 -5.1187799987E-09 -5.1187799987E-09
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
toldfe 1.00000000E-06 Hartree
tsmear11 1.00000000E-02 Hartree
tsmear12 2.00000000E-02 Hartree
tsmear13 3.00000000E-02 Hartree
tsmear14 4.00000000E-02 Hartree
tsmear21 1.00000000E-02 Hartree
tsmear22 2.00000000E-02 Hartree
tsmear23 3.00000000E-02 Hartree
tsmear24 4.00000000E-02 Hartree
tsmear31 1.00000000E-02 Hartree
tsmear32 2.00000000E-02 Hartree
tsmear33 3.00000000E-02 Hartree
tsmear34 4.00000000E-02 Hartree
typat 1
wtk11 0.75000 0.25000
wtk12 0.75000 0.25000
wtk13 0.75000 0.25000
wtk14 0.75000 0.25000
wtk21 0.09375 0.09375 0.09375 0.18750 0.09375 0.09375
0.09375 0.18750 0.03125 0.03125
wtk22 0.09375 0.09375 0.09375 0.18750 0.09375 0.09375
0.09375 0.18750 0.03125 0.03125
wtk23 0.09375 0.09375 0.09375 0.18750 0.09375 0.09375
0.09375 0.18750 0.03125 0.03125
wtk24 0.09375 0.09375 0.09375 0.18750 0.09375 0.09375
0.09375 0.18750 0.03125 0.03125
wtk31 0.02778 0.02778 0.02778 0.05556 0.02778 0.02778
0.05556 0.02778 0.05556 0.05556 0.02778 0.02778
0.02778 0.05556 0.02778 0.05556 0.05556 0.05556
0.02778 0.02778 0.02778 0.02778 0.02778 0.05556
0.05556 0.00926 0.00926 0.00926
wtk32 0.02778 0.02778 0.02778 0.05556 0.02778 0.02778
0.05556 0.02778 0.05556 0.05556 0.02778 0.02778
0.02778 0.05556 0.02778 0.05556 0.05556 0.05556
0.02778 0.02778 0.02778 0.02778 0.02778 0.05556
0.05556 0.00926 0.00926 0.00926
wtk33 0.02778 0.02778 0.02778 0.05556 0.02778 0.02778
0.05556 0.02778 0.05556 0.05556 0.02778 0.02778
0.02778 0.05556 0.02778 0.05556 0.05556 0.05556
0.02778 0.02778 0.02778 0.02778 0.02778 0.05556
0.05556 0.00926 0.00926 0.00926
wtk34 0.02778 0.02778 0.02778 0.05556 0.02778 0.02778
0.05556 0.02778 0.05556 0.05556 0.02778 0.02778
0.02778 0.05556 0.02778 0.05556 0.05556 0.05556
0.02778 0.02778 0.02778 0.02778 0.02778 0.05556
0.05556 0.00926 0.00926 0.00926
znucl 13.00000
================================================================================
- Timing analysis has been suppressed with timopt=0
================================================================================
Suggested references for the acknowledgment of ABINIT usage.
The users of ABINIT have little formal obligations with respect to the ABINIT group
(those specified in the GNU General Public License, http://www.gnu.org/copyleft/gpl.txt).
However, it is common practice in the scientific literature,
to acknowledge the efforts of people that have made the research possible.
In this spirit, please find below suggested citations of work written by ABINIT developers,
corresponding to implementations inside of ABINIT that you have used in the present run.
Note also that it will be of great value to readers of publications presenting these results,
to read papers enabling them to understand the theoretical formalism and details
of the ABINIT implementation.
For information on why they are suggested, see also https://docs.abinit.org/theory/acknowledgments.
-
- [1] Libxc: A library of exchange and correlation functionals for density functional theory.
- M.A.L. Marques, M.J.T. Oliveira, T. Burnus, Computer Physics Communications 183, 2227 (2012).
- Comment: to be cited when LibXC is used (negative value of ixc)
- Strong suggestion to cite this paper.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#marques2012
-
- [2] The Abinit project: Impact, environment and recent developments.
- Computer Phys. Comm. 248, 107042 (2020).
- X.Gonze, B. Amadon, G. Antonius, F.Arnardi, L.Baguet, J.-M.Beuken,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, N.Brouwer, F.Bruneval,
- G.Brunin, T.Cavignac, J.-B. Charraud, Wei Chen, M.Cote, S.Cottenier,
- J.Denier, G.Geneste, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, Xu He, N.Helbig, N.Holzwarth, Y.Jia, F.Jollet,
- W.Lafargue-Dit-Hauret, K.Lejaeghere, M.A.L.Marques, A.Martin, C.Martins,
- H.P.C. Miranda, F.Naccarato, K. Persson, G.Petretto, V.Planes, Y.Pouillon,
- S.Prokhorenko, F.Ricci, G.-M.Rignanese, A.H.Romero, M.M.Schmitt, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, G.Zerah and J.W.Zwanzig
- Comment: the fifth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT20.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2020
-
- [3] Optimized norm-conserving Vanderbilt pseudopotentials.
- D.R. Hamann, Phys. Rev. B 88, 085117 (2013).
- Comment: Some pseudopotential generated using the ONCVPSP code were used.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#hamann2013
-
- [4] ABINIT: Overview, and focus on selected capabilities
- J. Chem. Phys. 152, 124102 (2020).
- A. Romero, D.C. Allan, B. Amadon, G. Antonius, T. Applencourt, L.Baguet,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, F.Bruneval,
- G.Brunin, D.Caliste, M.Cote,
- J.Denier, C. Dreyer, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, F.Jollet, G. Jomard,
- A.Martin,
- H.P.C. Miranda, F.Naccarato, G.Petretto, N.A. Pike, V.Planes,
- S.Prokhorenko, T. Rangel, F.Ricci, G.-M.Rignanese, M.Royo, M.Stengel, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, J.Wiktor, J.W.Zwanziger, and X.Gonze.
- Comment: a global overview of ABINIT, with focus on selected capabilities .
- Note that a version of this paper, that is not formatted for J. Chem. Phys
- is available at https://www.abinit.org/sites/default/files/ABINIT20_JPC.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#romero2020
-
- [5] Recent developments in the ABINIT software package.
- Computer Phys. Comm. 205, 106 (2016).
- X.Gonze, F.Jollet, F.Abreu Araujo, D.Adams, B.Amadon, T.Applencourt,
- C.Audouze, J.-M.Beuken, J.Bieder, A.Bokhanchuk, E.Bousquet, F.Bruneval
- D.Caliste, M.Cote, F.Dahm, F.Da Pieve, M.Delaveau, M.Di Gennaro,
- B.Dorado, C.Espejo, G.Geneste, L.Genovese, A.Gerossier, M.Giantomassi,
- Y.Gillet, D.R.Hamann, L.He, G.Jomard, J.Laflamme Janssen, S.Le Roux,
- A.Levitt, A.Lherbier, F.Liu, I.Lukacevic, A.Martin, C.Martins,
- M.J.T.Oliveira, S.Ponce, Y.Pouillon, T.Rangel, G.-M.Rignanese,
- A.H.Romero, B.Rousseau, O.Rubel, A.A.Shukri, M.Stankovski, M.Torrent,
- M.J.Van Setten, B.Van Troeye, M.J.Verstraete, D.Waroquier, J.Wiktor,
- B.Xu, A.Zhou, J.W.Zwanziger.
- Comment: the fourth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT16.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2016
-
- Proc. 0 individual time (sec): cpu= 5.1 wall= 5.2
================================================================================
Calculation completed.
.Delivered 35 WARNINGs and 99 COMMENTs to log file.
+Overall time at end (sec) : cpu= 5.1 wall= 5.2
From the output file, here is the evolution of acell:
acell11 7.6022357792E+00 7.6022357792E+00 7.6022357792E+00 Bohr
acell12 7.6022341271E+00 7.6022341271E+00 7.6022341271E+00 Bohr
acell13 7.6022341214E+00 7.6022341214E+00 7.6022341214E+00 Bohr
acell14 7.6022357148E+00 7.6022357148E+00 7.6022357148E+00 Bohr
acell21 7.5604102145E+00 7.5604102145E+00 7.5604102145E+00 Bohr
acell22 7.5605496029E+00 7.5605496029E+00 7.5605496029E+00 Bohr
acell23 7.5565044147E+00 7.5565044147E+00 7.5565044147E+00 Bohr
acell24 7.5593333886E+00 7.5593333886E+00 7.5593333886E+00 Bohr
acell31 7.5483073963E+00 7.5483073963E+00 7.5483073963E+00 Bohr
acell32 7.5482393302E+00 7.5482393302E+00 7.5482393302E+00 Bohr
acell33 7.5497784006E+00 7.5497784006E+00 7.5497784006E+00 Bohr
acell34 7.5521340033E+00 7.5521340033E+00 7.5521340033E+00 Bohr
These data should be analyzed properly. For tsmear = 0.01, the converged value, contained in acell31, must be compared to acell11 and acell21: between acell21 and acell31, the difference is below 0.2%. acell31 can be considered to be converged with respect to the number of k-points, at fixed tsmear. This tsmear being the lowest one, it is usually the most difficult to converge, and the values acell31,32,33 and 34 are indeed well-converged with respect to the k-point number. The use of the largest tsmear = 0.04, giving acell34, induces only a small error in the lattice parameter. For that particular value of tsmear, one can use the second k-point grid, giving acell24.
Summary
So to summarize: we can choose to work with a 10 k-point grid in the irreducible Brillouin zone, and the associated tsmear = 0.04, with less than 0.1% error on the lattice parameter. Note that this error due to the Brillouin zone sampling could add to the error due to the choice of ecut (that was mentioned previously to be on the order of 0.2%).
In what follows, we will stick to these values of ecut and tsmear and try to use k-point grids with a similar resolution.
Our final value for the aluminum lattice parameter, in the LDA, using the Al.psp8 pseudopotential, is thus 7.5593 Bohr, which corresponds to 4.0002 Angstrom. The experimental value at 25 Celsius is 4.04958 Angstrom, hence our theoretical value has an error of 1.2%. We caution that converged parameters should be used to properly assess the accuracy of a pseudopotential and functional.
The associated total energy and precision can be deduced from:
etotal11 -2.3516656074E+00
etotal12 -2.3532597160E+00
etotal13 -2.3548538247E+00
etotal14 -2.3564479440E+00
etotal21 -2.3568282638E+00
etotal22 -2.3574128355E+00
etotal23 -2.3576771874E+00
etotal24 -2.3578584768E+00
etotal31 -2.3582092001E+00
etotal32 -2.3581800122E+00
etotal33 -2.3581917663E+00
etotal34 -2.3582884106E+00
etotal 24 is -2.3578584768E+00 Ha, with a precision of 0.0005 Ha.
Tip
To analyze the convergence of the total energy, one can use the abicomp.py script
provide by AbiPy and the gsr command that will start an interactive ipython session
so that we can interact directly with the AbiPy object.
To load all the GSR files produced by calculation, use the command
abicomp.py gsr tbase4_3o*GSR.nc
then, inside the ipython terminal, execute the plot_convergence method of the GsrRobot:
In [1]: robot.plot_convergence("energy", sortby="nkpt", hue="tsmear")
to produce this plot with the total energy in eV for different values of nkpt grouped by tsmear:
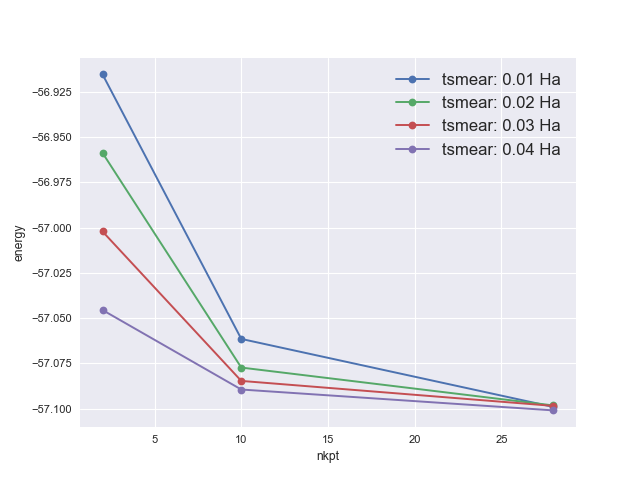
Surface energy of aluminum (100): changing the orientation of the unit cell¶
In order to study the Aluminum (100) surface, we will have to set up a supercell representing a slab.
This supercell should be chosen as to be compatible with the primitive surface unit cell.
The corresponding directions are [-1 1 0] and [1 1 0]. The direction perpendicular to the surface is [0 0 1].
There is no primitive cell of bulk aluminum based on these vectors, but a doubled cell.
We will first compute the total energy associated with this doubled cell.
This is not strictly needed, but it is a valuable intermediate step towards the study of the surface.
You might start from tbase4_3.abi. You have to change rprim. Still, try to keep acell at the values of bulk aluminum that were determined previously. But it is not all: the most difficult part in the passage to this doubled cell is the definition of the k-point grid. Of course, one could just take a homogeneous simple cubic grid of k-points, but this will not correspond exactly to the k-point grid used in the primitive cell in tbase4_3.abi. This would not be a big problem, but you would miss some error cancellation.
The answer to this problem is given in the input file $ABI_TESTS/tutorial/Input/tbase4_4.abi.
# Crystalline aluminum # # A first step in the determination of the surface energy of aluminum : # changing the orientation of the cell. #Definition of the unit cell acell 3*7.5593333886E+00 # Lattice parameters of bulk aluminum obtained # in the previous calculation. rprim 0.5 -0.5 0.0 # These values will define a cell with two atoms, 0.5 0.5 0.0 # non-primitive, with a different orientation 0.0 0.0 1.0 # than the primitive cell. chkprim 0 # This input variable allows to use non-primitive unit # cells. Please, do not use it in other cases, # you might miss a primitive cell, faster to handle. #Definition of the atom types ntypat 1 # There is only one type of atom znucl 13 # The keyword "znucl" refers to the atomic number of the # possible type(s) of atom. The pseudopotential(s) # mentioned in the "files" file must correspond # to the type(s) of atom. Here, the only type is Aluminum pp_dirpath "$ABI_PSPDIR" # This is the path to the directory were # pseudopotentials for tests are stored pseudos "Psdj_nc_sr_04_pw_std_psp8/Al.psp8" # Name and location of the pseudopotential #Definition of the atoms natom 2 # There are two atoms in this doubled cell typat 2*1 # These atoms are of type 1, that is, Aluminum xred # This keyword indicate that the location of the atoms # will follow, one triplet of number for each atom 0.0 0.0 0.0 # Triplet giving the REDUCED coordinate of atom 1. 0.5 0.5 0.5 # Triplet giving the REDUCED coordinate of atom 2. #Definition of the planewave basis set ecut 6.0 # Maximal kinetic energy cut-off, in Hartree #Definition of the k-point grids ngkpt 4 4 4 nshiftk 2 shiftk 0.5 0.0 0.5 0.0 0.5 0.5 #Definition of the SCF procedure nstep 10 # Maximal number of SCF cycles toldfe 1.0d-6 # This value is WAY TOO LARGE for most realistic studies of materials #Definition of occupation numbers occopt 4 tsmear 0.04 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% tbase4_4.abo, tolnlines= 7, tolabs= 2.008e-10, tolrel= 4.000e-06 #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = Unknown #%% keywords = #%% description = #%% Crystalline aluminum #%% #%% A first step in the determination of the surface energy of aluminum : #%% changing the orientation of the cell. #%% topics = UnitCell #%%<END TEST_INFO>
The procedure to do the exact translation of the k-point grid will not be explained here (sorry for this).
If you do not see how to do it, just use homogeneous simple cubic grids, with about the same resolution
as for the primitive cell case. There is a simple rule to estimate roughly whether two
grids for different cells have the same resolution: simply multiply the linear dimensions of the k-point grids,
by the number of sublattices, by the number of atoms in the cell.
For example, the corresponding product for the usual 10 k-point grid is 4x4x4 x 4 x 1 = 256.
In the file tbase4_4.in, one has 4x4x4 x 2 x 2 = 256.
The grids of k-points should not be too anisotropic for this rough estimation to be valid.
Note also the input variables rprim and chkprim in this input file.
Now run tbase4_4.abi (the reference file is $ABI_TESTS/tutorial/Refs/tbase4_4.abo). You should find the following total energy:
etotal -4.7164794308E+00
It is not exactly twice the total energy for the primitive cell, mentioned above, but the difference is less than 0.001 Ha. It is due to the different FFT grids used in the two runs, and affect the exchange-correlation energy. These grids are always homogeneous primitive 3D grids, so that changing the orientation of the lattice will give mutually incompatible lattices. Increasing the size of the FFT grid would improve the agreement.
Surface energy: a (3 aluminum layer + 1 vacuum layer) slab calculation¶
We will first compute the total energy associated with only three layers of aluminum, separated by only one layer of vacuum. This is kind of a minimal slab:
- one surface layer
- one “bulk” layer
- one surface layer
- one vacuum layer
- …
It is convenient to take the vacuum region as having a multiple of the width of the aluminum layers, but this is not mandatory.
The supercell to use is the double of the previous cell (that had two layers of Aluminum atoms along the [0 0 1] direction).
Of course, the relaxation of the surface might give an important contribution to the total energy.
You should start from tbase4_4.abi.
You have to modify rprim (double the cell along [0 0 1]), the atomic positions, as well as the k-point mesh.
For the latter, it is supposed that the electrons cannot propagate from one slab to its image in the [0 0 1] direction,
so that the \(k_z\) component of the special k-points can be taken 0: only one layer of k-points is needed along the z-direction.
You should also allow the relaxation of atomic positions, but not the relaxation of lattice parameters
(the lattice parameters along x or y must be considered fixed to the bulk value, while, for the z direction,
there is no interest to allow the vacuum region to collapse!
The input file tbase4_5.abi is an example,
# Crystalline aluminum # # Determination of the surface energy of aluminum : # a minimal slab. #Definition of the unit cell acell 3*7.5593333886E+00 # Lattice parameters of bulk aluminum rprim 0.5 -0.5 0.0 # The lattice vector along the z direction 0.5 0.5 0.0 # is doubled with respect to previous run. 0.0 0.0 2.0 # #Definition of the atom types ntypat 1 # There is only one type of atom znucl 13 # The keyword "znucl" refers to the atomic number of the # possible type(s) of atom. The pseudopotential(s) # mentioned in the "files" file must correspond # to the type(s) of atom. Here, the only type is Aluminum pp_dirpath "$ABI_PSPDIR" # This is the path to the directory were # pseudopotentials for tests are stored pseudos "Psdj_nc_sr_04_pw_std_psp8/Al.psp8" # Name and location of the pseudopotential #Definition of the atoms natom 3 # Three atoms per cell: three aluminum layers and some vacuum typat 3*1 # These atoms are of type 1, that is, Aluminum xred # This keyword indicate that the location of the atoms # will follow, one triplet of number for each atom 0.0 0.0 0.0 # Triplet giving the REDUCED coordinate of atom 1. 0.5 0.5 0.25 # Triplet giving the REDUCED coordinate of atom 2. 0.0 0.0 0.5 # Triplet giving the REDUCED coordinate of atom 3. # The z-coordinate of the vacuum layer is 0.75 . #Definition of the planewave basis set ecut 6.0 # Maximal kinetic energy cut-off, in Hartree #Definition of the k-point grids ngkpt 4 4 1 nshiftk 2 shiftk 0.5 0.0 0.0 0.0 0.5 0.0 #Definition of the SCF procedure nstep 10 # Maximal number of SCF cycles toldff 5.0d-5 # The reduced positions of the atoms are not uniquely determined by symmetry. # Thus, a better SCF stopping criterion than toldfe can be used. #Definition of occupation numbers occopt 4 tsmear 0.04 #The relaxation geoopt "bfgs" tolmxf 5.0d-4 # This might be not sufficiently converged. The default is more stringent. ntime 10 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% tbase4_5.abo, tolnlines= 0, tolabs= 0.000e+00, tolrel= 0.000e+00, fld_options=-easy #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = Unknown #%% keywords = #%% description = #%% Crystalline aluminum slab #%% #%% Determination of the surface energy of aluminum : #%% a minimal slab. #%%<END TEST_INFO>
while tbase4_5.abo is the reference output file.
.Version 10.5.8.2 of ABINIT, released Oct 2025.
.(MPI version, prepared for a x86_64_linux_gnu13.2 computer)
.Copyright (C) 1998-2026 ABINIT group .
ABINIT comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Sat 20 Dec 2025.
- ( at 17h06 )
- input file -> /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/TestBot_MPI1/tutorial_tbase4_5/tbase4_5.abi
- output file -> tbase4_5.abo
- root for input files -> tbase4_5i
- root for output files -> tbase4_5o
Symmetries : space group P4/m m m (#123); Bravais tP (primitive tetrag.)
================================================================================
Values of the parameters that define the memory need of the present run
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 36 mpssoang = 3 mqgrid = 3001
natom = 3 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 16 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 7 mffmem = 1 mkmem = 6
mpw = 316 nfft = 5184 nkpt = 6
================================================================================
P This job should need less than 3.375 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.205 Mbytes ; DEN or POT disk file : 0.042 Mbytes.
================================================================================
--------------------------------------------------------------------------------
------------- Echo of variables that govern the present computation ------------
--------------------------------------------------------------------------------
-
- outvars: echo of selected default values
- iomode0 = 0 , fftalg0 =512 , wfoptalg0 = 0
-
- outvars: echo of global parameters not present in the input file
- max_nthreads = 0
-
-outvars: echo values of preprocessed input variables --------
acell 7.5593333886E+00 7.5593333886E+00 7.5593333886E+00 Bohr
amu 2.69815390E+01
ecut 6.00000000E+00 Hartree
- fftalg 512
ionmov 2
ixc -1012
kpt -1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
-2.50000000E-01 1.25000000E-01 0.00000000E+00
5.00000000E-01 1.25000000E-01 0.00000000E+00
-3.75000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 3.75000000E-01 0.00000000E+00
kptrlatt 4 4 0 -4 4 0 0 0 1
kptrlen 1.51186668E+01
P mkmem 6
natom 3
nband 7
ngfft 12 12 36
nkpt 6
nstep 10
nsym 16
ntime 10
ntypat 1
occ 2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
occopt 4
optforces 1
rprim 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 2.0000000000E+00
shiftk 5.00000000E-01 5.00000000E-01 0.00000000E+00
spgroup 123
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
-1 0 0 0 1 0 0 0 -1 1 0 0 0 -1 0 0 0 1
-1 0 0 0 -1 0 0 0 1 1 0 0 0 1 0 0 0 -1
1 0 0 0 -1 0 0 0 -1 -1 0 0 0 1 0 0 0 1
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 -1 0 1 0 0 0 0 -1 0 1 0 -1 0 0 0 0 1
0 -1 0 -1 0 0 0 0 1 0 1 0 1 0 0 0 0 -1
0 1 0 -1 0 0 0 0 -1 0 -1 0 1 0 0 0 0 1
tnons 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.5000000
0.0000000 0.0000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.5000000
0.0000000 0.0000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.5000000
0.0000000 0.0000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.5000000
0.0000000 0.0000000 0.5000000 0.0000000 0.0000000 0.0000000
toldff 5.00000000E-05
tolmxf 5.00000000E-04
tsmear 4.00000000E-02 Hartree
typat 1 1 1
wtk 0.12500 0.12500 0.25000 0.12500 0.25000 0.12500
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
2.0001134781E+00 0.0000000000E+00 2.0001134781E+00
0.0000000000E+00 0.0000000000E+00 4.0002269562E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
3.7796666943E+00 0.0000000000E+00 3.7796666943E+00
0.0000000000E+00 0.0000000000E+00 7.5593333886E+00
xred 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 2.5000000000E-01
0.0000000000E+00 0.0000000000E+00 5.0000000000E-01
znucl 13.00000
================================================================================
chkinp: Checking input parameters for consistency.
================================================================================
== DATASET 1 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 1, }
dimensions: {natom: 3, nkpt: 6, mband: 7, nsppol: 1, nspinor: 1, nspden: 1, mpw: 316, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 9.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 4.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 0, iscf: 7, paral_kgb: 0, }
...
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 3.7796667 -3.7796667 0.0000000 G(1)= 0.1322868 -0.1322868 0.0000000
R(2)= 3.7796667 3.7796667 0.0000000 G(2)= 0.1322868 0.1322868 0.0000000
R(3)= 0.0000000 0.0000000 15.1186668 G(3)= 0.0000000 0.0000000 0.0661434
Unit cell volume ucvol= 4.3196693E+02 bohr^3
Angles (23,13,12)= 9.00000000E+01 9.00000000E+01 9.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 12 12 36
ecut(hartree)= 6.000 => boxcut(ratio)= 2.03597
--- Pseudopotential description ------------------------------------------------
- pspini: atom type 1 psp file is /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/Pspdir/Psdj_nc_sr_04_pw_std_psp8/Al.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/Pspdir/Psdj_nc_sr_04_pw_std_psp8/Al.psp8
- Al ONCVPSP-3.3.0 r_core= 1.76802 1.76802 1.70587
- 13.00000 3.00000 171102 znucl, zion, pspdat
8 -1012 2 4 600 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
5.99000000000000 5.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2 2
extension_switch 1
pspatm : epsatm= 0.57439192
--- l ekb(1:nproj) -->
0 5.725870 0.726131
1 6.190420 0.914022
2 -4.229503 -0.925599
pspatm: atomic psp has been read and splines computed
1.55085818E+01 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
_setup2: Arith. and geom. avg. npw (full set) are 303.750 303.688
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -6.9829579525527 -6.983E+00 1.250E-02 1.102E+02 4.724E-03 4.724E-03
ETOT 2 -7.0418462217034 -5.889E-02 1.873E-04 6.489E-01 7.955E-03 3.231E-03
ETOT 3 -7.0425335066752 -6.873E-04 1.647E-05 3.667E-01 4.259E-04 2.805E-03
ETOT 4 -7.0427092321560 -1.757E-04 3.372E-06 9.077E-03 3.573E-05 2.841E-03
ETOT 5 -7.0427135007667 -4.269E-06 3.571E-06 8.884E-05 3.591E-05 2.805E-03
At SCF step 5, forces are converged :
for the second time, max diff in force= 3.591E-05 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 6.52647296E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 6.52647296E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.73138514E-04 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 1, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 15.1186668, ]
lattice_lengths: [ 5.34526, 5.34526, 15.11867, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 4.3196693E+02
convergence: {deltae: -4.269E-06, res2: 8.884E-05, residm: 3.571E-06, diffor: 3.591E-05, }
etotal : -7.04271350E+00
entropy : 0.00000000E+00
fermie : 1.87742627E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 6.52647296E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 6.52647296E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 2.73138514E-04, ]
pressure_GPa: -3.9588E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
- [ 5.0000E-01, 5.0000E-01, 2.5000E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 5.0000E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -2.80519306E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, 2.80519306E-03, ]
force_length_stats: {min: 0.00000000E+00, max: 2.80519306E-03, mean: 1.87012871E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.88307009
2 2.00000 0.91422302
3 2.00000 0.88307009
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
3.77966669430000E+00 0.00000000000000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
5.00000000000000E-01 5.00000000000000E-01 2.50000000000000E-01
0.00000000000000E+00 0.00000000000000E+00 5.00000000000000E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 2.80519E-03 1.32238E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -2.80519306023203E-03
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 2.80519306023203E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 4.24107791233619E-02
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -4.24107791233619E-02
Total energy (etotal) [Ha]= -7.04271350076674E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -7.0427274822536 -7.043E+00 1.982E-06 2.840E-03 7.800E-05 2.727E-03
ETOT 2 -7.0427290336505 -1.551E-06 9.233E-07 5.346E-05 1.188E-05 2.715E-03
ETOT 3 -7.0427290769177 -4.327E-08 8.294E-07 2.719E-06 4.187E-06 2.720E-03
At SCF step 3, forces are converged :
for the second time, max diff in force= 4.187E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 6.61710574E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 6.61710574E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.75308579E-04 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 1, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 15.1186668, ]
lattice_lengths: [ 5.34526, 5.34526, 15.11867, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 4.3196693E+02
convergence: {deltae: -4.327E-08, res2: 2.719E-06, residm: 8.294E-07, diffor: 4.187E-06, }
etotal : -7.04272908E+00
entropy : 0.00000000E+00
fermie : 1.87769583E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 6.61710574E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 6.61710574E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 2.75308579E-04, ]
pressure_GPa: -3.9978E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -1.8555E-04, Al]
- [ 5.0000E-01, 5.0000E-01, 2.5000E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 5.0019E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -2.71950010E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, 2.71950010E-03, ]
force_length_stats: {min: 0.00000000E+00, max: 2.71950010E-03, mean: 1.81300007E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.88305130
2 2.00000 0.91379400
3 2.00000 0.88305130
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -2.80519306023203E-03
3.77966669430000E+00 0.00000000000000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.56213858166023E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -1.85545002186466E-04
5.00000000000000E-01 5.00000000000000E-01 2.50000000000000E-01
0.00000000000000E+00 0.00000000000000E+00 5.00185545002186E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 2.71950E-03 1.28198E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -2.71950010390740E-03
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 2.71950010390740E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 4.11152158715367E-02
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -4.11152158715367E-02
Total energy (etotal) [Ha]= -7.04272907691768E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.55762E-05
Relative =-2.21167E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -7.0428384397157 -7.043E+00 1.563E-05 2.121E-01 2.781E-03 6.104E-05
ETOT 2 -7.0429717863609 -1.333E-04 3.734E-07 1.619E-02 1.265E-04 6.547E-05
ETOT 3 -7.0429806371782 -8.851E-06 8.203E-08 4.407E-05 6.034E-05 1.258E-04
ETOT 4 -7.0429806824510 -4.527E-08 5.302E-08 6.380E-06 3.444E-06 1.293E-04
ETOT 5 -7.0429806855646 -3.114E-09 2.276E-09 2.710E-07 3.232E-07 1.296E-04
At SCF step 5, forces are converged :
for the second time, max diff in force= 3.232E-07 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 8.93284299E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 8.93284299E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.19575613E-04 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 1, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 15.1186668, ]
lattice_lengths: [ 5.34526, 5.34526, 15.11867, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 4.3196693E+02
convergence: {deltae: -3.114E-09, res2: 2.710E-07, residm: 2.276E-09, diffor: 3.232E-07, }
etotal : -7.04298069E+00
entropy : 0.00000000E+00
fermie : 1.89624295E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 8.93284299E-05, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 8.93284299E-05, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.19575613E-04, ]
pressure_GPa: -4.8862E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -6.0739E-03, Al]
- [ 5.0000E-01, 5.0000E-01, 2.5000E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 5.0607E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -1.29577871E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, 1.29577871E-04, ]
force_length_stats: {min: 0.00000000E+00, max: 1.29577871E-04, mean: 8.63852471E-05, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.88800434
2 2.00000 0.90258245
3 2.00000 0.88800434
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -9.18291122477249E-02
3.77966669430000E+00 0.00000000000000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.65116250084773E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -6.07388955659831E-03
5.00000000000000E-01 5.00000000000000E-01 2.50000000000000E-01
0.00000000000000E+00 0.00000000000000E+00 5.06073889556599E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 1.29578E-04 6.10836E-05 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -1.29577870673370E-04
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 1.29577870673370E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 1.95904464840980E-03
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -1.95904464840980E-03
Total energy (etotal) [Ha]= -7.04298068556457E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.51609E-04
Relative =-3.57254E-05
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 1.2958E-04 < tolmxf= 5.0000E-04 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 67.822E-12; max= 22.764E-10
reduced coordinates (array xred) for 3 atoms
0.000000000000 0.000000000000 -0.006073889557
0.500000000000 0.500000000000 0.250000000000
0.000000000000 0.000000000000 0.506073889557
rms dE/dt= 9.2350E-04; max dE/dt= 1.9590E-03; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.001959044648
2 0.000000000000 0.000000000000 0.000000000000
3 0.000000000000 0.000000000000 -0.001959044648
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 -0.04859387346598
2 2.00011347807574 0.00000000000000 2.00011347807574
3 0.00000000000000 0.00000000000000 4.04882082961746
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00012957787067
2 -0.00000000000000 -0.00000000000000 -0.00000000000000
3 -0.00000000000000 -0.00000000000000 0.00012957787067
frms,max,avg= 6.1083594E-05 1.2957787E-04 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00666316201372
2 -0.00000000000000 -0.00000000000000 -0.00000000000000
3 -0.00000000000000 -0.00000000000000 0.00666316201372
frms,max,avg= 3.1410447E-03 6.6631620E-03 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.559333388600 7.559333388600 7.559333388600 bohr
= 4.000226956151 4.000226956151 4.000226956151 angstroms
prteigrs : about to open file tbase4_5o_EIG
Fermi (or HOMO) energy (hartree) = 0.18962 Average Vxc (hartree)= -0.32633
Eigenvalues (hartree) for nkpt= 6 k points:
kpt# 1, nband= 7, wtk= 0.12500, kpt= -0.1250 0.0000 0.0000 (reduced coord)
-0.18867 -0.10186 -0.00740 0.16603 0.24052 0.29539 0.37431
occupation numbers for kpt# 1
2.00000 2.00000 2.00000 1.79676 0.00020 0.00071 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 1, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 2.40908590605293E+00
hartree : 9.10290904527904E-01
xc : -3.23773398528802E+00
Ewald energy : -6.41005844417523E+00
psp_core : 3.59022435039527E-02
local_psp : -2.02757162433414E+00
non_local_psp : 1.27873706215166E+00
internal : -7.04134793756095E+00
'-kT*entropy' : -1.63274800361497E-03
total_energy : -7.04298068556457E+00
total_energy_eV : -1.91649267757881E+02
band_energy : 3.35636266920485E-01
...
rms coord change= 2.8633E-03 atom, delta coord (reduced):
1 0.000000000000 0.000000000000 -0.006073889557
2 0.000000000000 0.000000000000 0.000000000000
3 0.000000000000 0.000000000000 0.006073889557
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 8.93284299E-05 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 8.93284299E-05 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.19575613E-04 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= -4.8862E+00 GPa]
- sigma(1 1)= 2.62813314E+00 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= 2.62813314E+00 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= 9.40223914E+00 sigma(2 1)= 0.00000000E+00
== END DATASET(S) ==============================================================
================================================================================
-outvars: echo values of variables after computation --------
acell 7.5593333886E+00 7.5593333886E+00 7.5593333886E+00 Bohr
amu 2.69815390E+01
ecut 6.00000000E+00 Hartree
etotal -7.0429806856E+00
fcart -0.0000000000E+00 -0.0000000000E+00 -1.2957787067E-04
-0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
-0.0000000000E+00 -0.0000000000E+00 1.2957787067E-04
- fftalg 512
ionmov 2
ixc -1012
kpt -1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
-2.50000000E-01 1.25000000E-01 0.00000000E+00
5.00000000E-01 1.25000000E-01 0.00000000E+00
-3.75000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 3.75000000E-01 0.00000000E+00
kptrlatt 4 4 0 -4 4 0 0 0 1
kptrlen 1.51186668E+01
P mkmem 6
natom 3
nband 7
ngfft 12 12 36
nkpt 6
nstep 10
nsym 16
ntime 10
ntypat 1
occ 2.000000 2.000000 2.000000 1.796764 0.000200 0.000711
0.000000
2.000000 2.000000 2.000112 2.000959 2.140242 0.000009
0.001214
2.000000 2.000000 2.000002 1.394738 0.318811 0.005256
0.002585
2.000000 2.000000 2.000153 2.000243 2.140960 1.428201
0.000240
2.000000 2.000000 2.013016 2.052141 1.252250 0.004911
0.003351
2.000063 2.000129 2.141840 2.141830 0.071186 0.037294
0.003529
occopt 4
optforces 1
rprim 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 2.0000000000E+00
shiftk 5.00000000E-01 5.00000000E-01 0.00000000E+00
spgroup 123
strten 8.9328429932E-05 8.9328429932E-05 3.1957561269E-04
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
-1 0 0 0 1 0 0 0 -1 1 0 0 0 -1 0 0 0 1
-1 0 0 0 -1 0 0 0 1 1 0 0 0 1 0 0 0 -1
1 0 0 0 -1 0 0 0 -1 -1 0 0 0 1 0 0 0 1
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 -1 0 1 0 0 0 0 -1 0 1 0 -1 0 0 0 0 1
0 -1 0 -1 0 0 0 0 1 0 1 0 1 0 0 0 0 -1
0 1 0 -1 0 0 0 0 -1 0 -1 0 1 0 0 0 0 1
tnons 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.5000000
0.0000000 0.0000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.5000000
0.0000000 0.0000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.5000000
0.0000000 0.0000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.5000000
0.0000000 0.0000000 0.5000000 0.0000000 0.0000000 0.0000000
toldff 5.00000000E-05
tolmxf 5.00000000E-04
tsmear 4.00000000E-02 Hartree
typat 1 1 1
wtk 0.12500 0.12500 0.25000 0.12500 0.25000 0.12500
xangst 0.0000000000E+00 0.0000000000E+00 -4.8593873466E-02
2.0001134781E+00 0.0000000000E+00 2.0001134781E+00
0.0000000000E+00 0.0000000000E+00 4.0488208296E+00
xcart 0.0000000000E+00 0.0000000000E+00 -9.1829112248E-02
3.7796666943E+00 0.0000000000E+00 3.7796666943E+00
0.0000000000E+00 0.0000000000E+00 7.6511625008E+00
xred 0.0000000000E+00 0.0000000000E+00 -6.0738895566E-03
5.0000000000E-01 5.0000000000E-01 2.5000000000E-01
0.0000000000E+00 0.0000000000E+00 5.0607388956E-01
znucl 13.00000
================================================================================
- Timing analysis has been suppressed with timopt=0
================================================================================
Suggested references for the acknowledgment of ABINIT usage.
The users of ABINIT have little formal obligations with respect to the ABINIT group
(those specified in the GNU General Public License, http://www.gnu.org/copyleft/gpl.txt).
However, it is common practice in the scientific literature,
to acknowledge the efforts of people that have made the research possible.
In this spirit, please find below suggested citations of work written by ABINIT developers,
corresponding to implementations inside of ABINIT that you have used in the present run.
Note also that it will be of great value to readers of publications presenting these results,
to read papers enabling them to understand the theoretical formalism and details
of the ABINIT implementation.
For information on why they are suggested, see also https://docs.abinit.org/theory/acknowledgments.
-
- [1] Libxc: A library of exchange and correlation functionals for density functional theory.
- M.A.L. Marques, M.J.T. Oliveira, T. Burnus, Computer Physics Communications 183, 2227 (2012).
- Comment: to be cited when LibXC is used (negative value of ixc)
- Strong suggestion to cite this paper.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#marques2012
-
- [2] The Abinit project: Impact, environment and recent developments.
- Computer Phys. Comm. 248, 107042 (2020).
- X.Gonze, B. Amadon, G. Antonius, F.Arnardi, L.Baguet, J.-M.Beuken,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, N.Brouwer, F.Bruneval,
- G.Brunin, T.Cavignac, J.-B. Charraud, Wei Chen, M.Cote, S.Cottenier,
- J.Denier, G.Geneste, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, Xu He, N.Helbig, N.Holzwarth, Y.Jia, F.Jollet,
- W.Lafargue-Dit-Hauret, K.Lejaeghere, M.A.L.Marques, A.Martin, C.Martins,
- H.P.C. Miranda, F.Naccarato, K. Persson, G.Petretto, V.Planes, Y.Pouillon,
- S.Prokhorenko, F.Ricci, G.-M.Rignanese, A.H.Romero, M.M.Schmitt, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, G.Zerah and J.W.Zwanzig
- Comment: the fifth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT20.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2020
-
- [3] Optimized norm-conserving Vanderbilt pseudopotentials.
- D.R. Hamann, Phys. Rev. B 88, 085117 (2013).
- Comment: Some pseudopotential generated using the ONCVPSP code were used.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#hamann2013
-
- [4] ABINIT: Overview, and focus on selected capabilities
- J. Chem. Phys. 152, 124102 (2020).
- A. Romero, D.C. Allan, B. Amadon, G. Antonius, T. Applencourt, L.Baguet,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, F.Bruneval,
- G.Brunin, D.Caliste, M.Cote,
- J.Denier, C. Dreyer, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, F.Jollet, G. Jomard,
- A.Martin,
- H.P.C. Miranda, F.Naccarato, G.Petretto, N.A. Pike, V.Planes,
- S.Prokhorenko, T. Rangel, F.Ricci, G.-M.Rignanese, M.Royo, M.Stengel, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, J.Wiktor, J.W.Zwanziger, and X.Gonze.
- Comment: a global overview of ABINIT, with focus on selected capabilities .
- Note that a version of this paper, that is not formatted for J. Chem. Phys
- is available at https://www.abinit.org/sites/default/files/ABINIT20_JPC.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#romero2020
-
- [5] Recent developments in the ABINIT software package.
- Computer Phys. Comm. 205, 106 (2016).
- X.Gonze, F.Jollet, F.Abreu Araujo, D.Adams, B.Amadon, T.Applencourt,
- C.Audouze, J.-M.Beuken, J.Bieder, A.Bokhanchuk, E.Bousquet, F.Bruneval
- D.Caliste, M.Cote, F.Dahm, F.Da Pieve, M.Delaveau, M.Di Gennaro,
- B.Dorado, C.Espejo, G.Geneste, L.Genovese, A.Gerossier, M.Giantomassi,
- Y.Gillet, D.R.Hamann, L.He, G.Jomard, J.Laflamme Janssen, S.Le Roux,
- A.Levitt, A.Lherbier, F.Liu, I.Lukacevic, A.Martin, C.Martins,
- M.J.T.Oliveira, S.Ponce, Y.Pouillon, T.Rangel, G.-M.Rignanese,
- A.H.Romero, B.Rousseau, O.Rubel, A.A.Shukri, M.Stankovski, M.Torrent,
- M.J.Van Setten, B.Van Troeye, M.J.Verstraete, D.Waroquier, J.Wiktor,
- B.Xu, A.Zhou, J.W.Zwanziger.
- Comment: the fourth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT16.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2016
-
- Proc. 0 individual time (sec): cpu= 2.0 wall= 2.0
================================================================================
Calculation completed.
.Delivered 2 WARNINGs and 2 COMMENTs to log file.
+Overall time at end (sec) : cpu= 2.0 wall= 2.0
The run will take a few second on a modern PC.
The total energy after the first SCF cycle, when the atomic positions are equal to their starting values, is:
ETOT 5 -7.0427135007667
The total energy of three aluminum atoms in the bulk, (from section 4.3, etotal24 multiplied by three) is -7.0735754304 Ha. Comparing the non-relaxed slab energy and the bulk energy, one obtains the non-relaxed surface energy, per surface unit cell (there are two surfaces in our simulation cell!), namely 0.01543 Ha = 0.420 eV.
The total energy after the Broyden relaxation is:
etotal -7.0429806856E+00
The relaxed surface energy, per surface unit cell, is obtained by comparing the bulk energy and the relaxed slab energy, and gives 0.015297 Ha = 0.416 eV. It seems that the relaxation energy is very small, compared to the surface energy, but we need to do the convergence studies.
Surface energy: increasing the number of vacuum layers¶
One should now increase the number of vacuum layers: 2 and 3 layers instead of only 1. It is preferable to define atomic positions in Cartesian coordinates. The same coordinates will work for both 2 and 3 vacuum layers, while this is not the case for reduced coordinates, as the cell size increases.
The input file tbase4_6.abi is an example input file,
# Crystalline aluminum : computation of the total energy # # Determination of the surface energy of aluminum : # convergence with respect to the number of vacuum layers. ndtset 2 #Definition of the unit cell acell 3*7.5593333886E+00 # Lattice parameters of bulk aluminum rprim1 0.5 -0.5 0.0 0.5 0.5 0.0 0.0 0.0 2.5 rprim2 0.5 -0.5 0.0 0.5 0.5 0.0 0.0 0.0 3.0 #Definition of the atom types ntypat 1 # There is only one type of atom znucl 13 # The keyword "znucl" refers to the atomic number of the # possible type(s) of atom. The pseudopotential(s) # mentioned in the "files" file must correspond # to the type(s) of atom. Here, the only type is Aluminum pp_dirpath "$ABI_PSPDIR" # This is the path to the directory were # pseudopotentials for tests are stored pseudos "Psdj_nc_sr_04_pw_std_psp8/Al.psp8" # Name and location of the pseudopotential #Definition of the atoms natom 3 # Three atoms per cell: three aluminum layers and some vacuum typat 3*1 # These atoms are of type 1, that is, Aluminum xcart 3*0.0 # Triplet giving the CARTESIAN coordinates of atom 1. 0.0 2*3.7796666943 # Triplet giving the CARTESIAN coordinates of atom 2. 2*0.0 7.5593333886 # Triplet giving the CARTESIAN coordinates of atom 3. #Definition of the planewave basis set ecut 6.0 # Maximal kinetic energy cut-off, in Hartree #Definition of the k-point grids ngkpt 4 4 1 nshiftk 2 shiftk 0.5 0.0 0.0 0.0 0.5 0.0 #Definition of the SCF procedure nstep 6 # Maximal number of SCF cycles toldff 5.0d-5 #Definition of occupation numbers occopt 4 tsmear 0.04 #The relaxation geoopt "bfgs" tolmxf 5.0d-4 ntime 10 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% tbase4_6.abo, tolnlines = 0, tolabs = 0.0e+00, tolrel = 0.0e+00, fld_options = -easy #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = Unknown #%% keywords = #%% description = #%% Crystalline aluminum : computation of the total energy #%% #%% Determination of the surface energy of aluminum : #%% convergence with respect to the number of vacuum layers. #%%<END TEST_INFO>
while tbase4_6.abo is the reference output file.
.Version 10.5.8.2 of ABINIT, released Oct 2025.
.(MPI version, prepared for a x86_64_linux_gnu13.2 computer)
.Copyright (C) 1998-2026 ABINIT group .
ABINIT comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Sat 20 Dec 2025.
- ( at 17h06 )
- input file -> /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/TestBot_MPI1/tutorial_tbase4_6/tbase4_6.abi
- output file -> tbase4_6.abo
- root for input files -> tbase4_6i
- root for output files -> tbase4_6o
DATASET 1 : space group P4/m m m (#123); Bravais tP (primitive tetrag.)
================================================================================
Values of the parameters that define the memory need for DATASET 1.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 45 mpssoang = 3 mqgrid = 3001
natom = 3 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 16 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 7 mffmem = 1 mkmem = 6
mpw = 388 nfft = 6480 nkpt = 6
================================================================================
P This job should need less than 3.847 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.251 Mbytes ; DEN or POT disk file : 0.051 Mbytes.
================================================================================
DATASET 2 : space group P4/m m m (#123); Bravais tP (primitive tetrag.)
================================================================================
Values of the parameters that define the memory need for DATASET 2.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 54 mpssoang = 3 mqgrid = 3001
natom = 3 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 16 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 7 mffmem = 1 mkmem = 6
mpw = 472 nfft = 7776 nkpt = 6
================================================================================
P This job should need less than 4.329 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.304 Mbytes ; DEN or POT disk file : 0.061 Mbytes.
================================================================================
--------------------------------------------------------------------------------
------------- Echo of variables that govern the present computation ------------
--------------------------------------------------------------------------------
-
- outvars: echo of selected default values
- iomode0 = 0 , fftalg0 =512 , wfoptalg0 = 0
-
- outvars: echo of global parameters not present in the input file
- max_nthreads = 0
-
-outvars: echo values of preprocessed input variables --------
acell 7.5593333886E+00 7.5593333886E+00 7.5593333886E+00 Bohr
amu 2.69815390E+01
ecut 6.00000000E+00 Hartree
- fftalg 512
ionmov 2
ixc -1012
jdtset 1 2
kpt -1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
-2.50000000E-01 1.25000000E-01 0.00000000E+00
5.00000000E-01 1.25000000E-01 0.00000000E+00
-3.75000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 3.75000000E-01 0.00000000E+00
kptrlatt 4 4 0 -4 4 0 0 0 1
kptrlen 3.02373336E+01
P mkmem 6
natom 3
nband 7
ndtset 2
ngfft1 12 12 45
ngfft2 12 12 54
nkpt 6
nstep 6
nsym 16
ntime 10
ntypat 1
occ 2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
occopt 4
optforces 1
rprim1 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 2.5000000000E+00
rprim2 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 3.0000000000E+00
shiftk 5.00000000E-01 5.00000000E-01 0.00000000E+00
spgroup 123
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
-1 0 0 0 1 0 0 0 -1 1 0 0 0 -1 0 0 0 1
-1 0 0 0 -1 0 0 0 1 1 0 0 0 1 0 0 0 -1
1 0 0 0 -1 0 0 0 -1 -1 0 0 0 1 0 0 0 1
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 -1 0 1 0 0 0 0 -1 0 1 0 -1 0 0 0 0 1
0 -1 0 -1 0 0 0 0 1 0 1 0 1 0 0 0 0 -1
0 1 0 -1 0 0 0 0 -1 0 -1 0 1 0 0 0 0 1
tnons1 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
tnons2 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.3333333
0.0000000 0.0000000 0.3333333 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.3333333
0.0000000 0.0000000 0.3333333 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.3333333
0.0000000 0.0000000 0.3333333 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.3333333
0.0000000 0.0000000 0.3333333 0.0000000 0.0000000 0.0000000
toldff 5.00000000E-05
tolmxf 5.00000000E-04
tsmear 4.00000000E-02 Hartree
typat 1 1 1
wtk 0.12500 0.12500 0.25000 0.12500 0.25000 0.12500
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 2.0001134781E+00 2.0001134781E+00
0.0000000000E+00 0.0000000000E+00 4.0002269562E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 3.7796666943E+00 3.7796666943E+00
0.0000000000E+00 0.0000000000E+00 7.5593333886E+00
xred1 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
-5.0000000000E-01 5.0000000000E-01 2.0000000000E-01
0.0000000000E+00 0.0000000000E+00 4.0000000000E-01
xred2 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
-5.0000000000E-01 5.0000000000E-01 1.6666666667E-01
0.0000000000E+00 0.0000000000E+00 3.3333333333E-01
znucl 13.00000
================================================================================
chkinp: Checking input parameters for consistency, jdtset= 1.
chkinp: Checking input parameters for consistency, jdtset= 2.
================================================================================
== DATASET 1 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 1, }
dimensions: {natom: 3, nkpt: 6, mband: 7, nsppol: 1, nspinor: 1, nspden: 1, mpw: 388, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 9.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 4.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 0, iscf: 7, paral_kgb: 0, }
...
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 3.7796667 -3.7796667 0.0000000 G(1)= 0.1322868 -0.1322868 0.0000000
R(2)= 3.7796667 3.7796667 0.0000000 G(2)= 0.1322868 0.1322868 0.0000000
R(3)= 0.0000000 0.0000000 18.8983335 G(3)= 0.0000000 0.0000000 0.0529147
Unit cell volume ucvol= 5.3995866E+02 bohr^3
Angles (23,13,12)= 9.00000000E+01 9.00000000E+01 9.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 12 12 45
ecut(hartree)= 6.000 => boxcut(ratio)= 2.03597
--- Pseudopotential description ------------------------------------------------
- pspini: atom type 1 psp file is /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/Pspdir/Psdj_nc_sr_04_pw_std_psp8/Al.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/Pspdir/Psdj_nc_sr_04_pw_std_psp8/Al.psp8
- Al ONCVPSP-3.3.0 r_core= 1.76802 1.76802 1.70587
- 13.00000 3.00000 171102 znucl, zion, pspdat
8 -1012 2 4 600 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
5.99000000000000 5.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2 2
extension_switch 1
pspatm : epsatm= 0.57439192
--- l ekb(1:nproj) -->
0 5.725870 0.726131
1 6.190420 0.914022
2 -4.229503 -0.925599
pspatm: atomic psp has been read and splines computed
1.55085818E+01 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
_setup2: Arith. and geom. avg. npw (full set) are 379.250 379.160
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 6, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -6.9582155601713 -6.958E+00 1.194E-02 1.964E+02 6.735E-03 6.735E-03
ETOT 2 -7.0333636484884 -7.515E-02 5.388E-03 3.262E+00 1.124E-02 4.500E-03
ETOT 3 -7.0338013653791 -4.377E-04 9.153E-05 4.651E+00 5.572E-04 3.943E-03
ETOT 4 -7.0348230859286 -1.022E-03 6.981E-05 8.674E-01 3.863E-04 4.330E-03
ETOT 5 -7.0350099634525 -1.869E-04 1.041E-05 3.055E-02 1.691E-04 4.499E-03
ETOT 6 -7.0350152828531 -5.319E-06 1.194E-06 3.441E-05 6.859E-05 4.567E-03
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.50656322E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.50656322E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -3.69136808E-05 sigma(2 1)= 0.00000000E+00
scprqt: WARNING -
nstep= 6 was not enough SCF cycles to converge;
maximum force difference= 6.859E-05 exceeds toldff= 5.000E-05
--- !ResultsGS
iteration_state: {dtset: 1, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 18.8983335, ]
lattice_lengths: [ 5.34526, 5.34526, 18.89833, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 5.3995866E+02
convergence: {deltae: -5.319E-06, res2: 3.441E-05, residm: 1.194E-06, diffor: 6.859E-05, }
etotal : -7.03501528E+00
entropy : 0.00000000E+00
fermie : 1.11495203E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.50656322E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.50656322E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -3.69136808E-05, ]
pressure_GPa: -2.5930E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
- [ -5.0000E-01, 5.0000E-01, 2.0000E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 4.0000E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -4.56718502E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -2.89120579E-19, ]
- [ -0.00000000E+00, -0.00000000E+00, 4.56718502E-03, ]
force_length_stats: {min: 2.89120579E-19, max: 4.56718502E-03, mean: 3.04479001E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.88599346
2 2.00000 0.91707854
3 2.00000 0.88599346
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-5.00000000000000E-01 5.00000000000000E-01 2.00000000000000E-01
0.00000000000000E+00 0.00000000000000E+00 4.00000000000000E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 4.56719E-03 2.15299E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -4.56718502091451E-03
-0.00000000000000E+00 -0.00000000000000E+00 -2.89120579329468E-19
-0.00000000000000E+00 -0.00000000000000E+00 4.56718502091451E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 8.63121855512821E-02
-0.00000000000000E+00 0.00000000000000E+00 5.46389712164155E-18
-0.00000000000000E+00 -0.00000000000000E+00 -8.63121855512821E-02
Total energy (etotal) [Ha]= -7.03501528285313E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 6, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -7.0350561703769 -7.035E+00 7.086E-08 4.071E-04 1.252E-04 4.442E-03
ETOT 2 -7.0350564312999 -2.609E-07 1.626E-09 2.402E-04 2.447E-06 4.444E-03
ETOT 3 -7.0350564833476 -5.205E-08 2.150E-10 5.361E-05 5.030E-06 4.449E-03
At SCF step 3, forces are converged :
for the second time, max diff in force= 5.030E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.51810186E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.51810186E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -3.61132200E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 1, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 18.8983335, ]
lattice_lengths: [ 5.34526, 5.34526, 18.89833, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 5.3995866E+02
convergence: {deltae: -5.205E-08, res2: 5.361E-05, residm: 2.150E-10, diffor: 5.030E-06, }
etotal : -7.03505648E+00
entropy : 0.00000000E+00
fermie : 1.11386989E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.51810186E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.51810186E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -3.61132200E-05, ]
pressure_GPa: -2.6234E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -2.4167E-04, Al]
- [ -5.0000E-01, 5.0000E-01, 2.0000E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 4.0024E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -4.44942422E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, 4.44942422E-03, ]
force_length_stats: {min: 0.00000000E+00, max: 4.44942422E-03, mean: 2.96628282E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.88613795
2 2.00000 0.91647708
3 2.00000 0.88613795
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -4.56718502091451E-03
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.56390057362092E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -2.41671310742936E-04
-5.00000000000000E-01 5.00000000000000E-01 2.00000000000000E-01
0.00000000000000E+00 0.00000000000000E+00 4.00241671310743E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 4.44942E-03 2.09748E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -4.44942422432432E-03
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 4.44942422432432E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 8.40867027474513E-02
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -8.40867027474513E-02
Total energy (etotal) [Ha]= -7.03505648334763E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-4.12005E-05
Relative =-5.85647E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 6, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -7.0353588104665 -7.035E+00 4.973E-06 1.002E+00 4.419E-03 3.022E-05
ETOT 2 -7.0358364433143 -4.776E-04 1.951E-07 1.007E-01 1.324E-04 1.626E-04
ETOT 3 -7.0358638335956 -2.739E-05 1.576E-07 1.323E-02 1.046E-04 2.672E-04
ETOT 4 -7.0358640306565 -1.971E-07 9.643E-09 1.008E-02 1.625E-05 2.835E-04
ETOT 5 -7.0358659542069 -1.924E-06 2.545E-08 1.671E-03 1.277E-05 2.962E-04
At SCF step 5, forces are converged :
for the second time, max diff in force= 1.277E-05 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.92487339E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.92487339E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 1.65359965E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 1, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 18.8983335, ]
lattice_lengths: [ 5.34526, 5.34526, 18.89833, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 5.3995866E+02
convergence: {deltae: -1.924E-06, res2: 1.671E-03, residm: 2.545E-08, diffor: 1.277E-05, }
etotal : -7.03586595E+00
entropy : 0.00000000E+00
fermie : 1.13392501E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.92487339E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.92487339E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 1.65359965E-05, ]
pressure_GPa: -3.9376E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -9.3729E-03, Al]
- [ -5.0000E-01, 5.0000E-01, 2.0000E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 4.0937E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -2.96237551E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, 2.96237551E-04, ]
force_length_stats: {min: 0.00000000E+00, max: 2.96237551E-04, mean: 1.97491700E-04, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.87826483
2 2.00000 0.89580335
3 2.00000 0.87826483
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -1.77131775762834E-01
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.73646516436283E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -9.37287809160319E-03
-5.00000000000000E-01 5.00000000000000E-01 2.00000000000000E-01
0.00000000000000E+00 0.00000000000000E+00 4.09372878091603E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 2.96238E-04 1.39648E-04 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -2.96237550583094E-04
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 2.96237550583094E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 5.59839601769965E-03
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -5.59839601769965E-03
Total energy (etotal) [Ha]= -7.03586595420693E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-8.09471E-04
Relative =-1.15056E-04
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 2.9624E-04 < tolmxf= 5.0000E-04 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 26.492E-10; max= 25.451E-09
reduced coordinates (array xred) for 3 atoms
0.000000000000 0.000000000000 -0.009372878092
-0.500000000000 0.500000000000 0.200000000000
0.000000000000 0.000000000000 0.409372878092
rms dE/dt= 2.6391E-03; max dE/dt= 5.5984E-03; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.005598396018
2 0.000000000000 0.000000000000 0.000000000000
3 0.000000000000 0.000000000000 -0.005598396018
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 -0.09373409899688
2 0.00000000000000 2.00011347807574 2.00011347807574
3 0.00000000000000 0.00000000000000 4.09396105514835
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00029623755058
2 -0.00000000000000 -0.00000000000000 -0.00000000000000
3 -0.00000000000000 -0.00000000000000 0.00029623755058
frms,max,avg= 1.3964772E-04 2.9623755E-04 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.01523314732544
2 -0.00000000000000 -0.00000000000000 -0.00000000000000
3 -0.00000000000000 -0.00000000000000 0.01523314732544
frms,max,avg= 7.1809745E-03 1.5233147E-02 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.559333388600 7.559333388600 7.559333388600 bohr
= 4.000226956151 4.000226956151 4.000226956151 angstroms
prteigrs : about to open file tbase4_6o_DS1_EIG
Fermi (or HOMO) energy (hartree) = 0.11339 Average Vxc (hartree)= -0.28254
Eigenvalues (hartree) for nkpt= 6 k points:
kpt# 1, nband= 7, wtk= 0.12500, kpt= -0.1250 0.0000 0.0000 (reduced coord)
-0.25238 -0.17285 -0.05780 0.05957 0.17268 0.23093 0.30445
occupation numbers for kpt# 1
2.00000 2.00000 2.00000 2.13526 0.00307 0.00020 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 1, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 2.38652637308975E+00
hartree : 3.31388209589652E+00
xc : -3.22004796346553E+00
Ewald energy : -3.39962709367289E+00
psp_core : 2.87217948031621E-02
local_psp : -7.42771255710694E+00
non_local_psp : 1.28372018565627E+00
internal : -7.03453716479964E+00
'-kT*entropy' : -1.32878940728794E-03
total_energy : -7.03586595420693E+00
total_energy_eV : -1.91455666054872E+02
band_energy : -2.54419531653270E-01
...
rms coord change= 4.4184E-03 atom, delta coord (reduced):
1 0.000000000000 0.000000000000 -0.009372878092
2 0.000000000000 0.000000000000 0.000000000000
3 0.000000000000 0.000000000000 0.009372878092
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.92487339E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.92487339E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 1.65359965E-05 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= -3.9376E+00 GPa]
- sigma(1 1)= 5.66317303E+00 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= 5.66317303E+00 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= 4.86505814E-01 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 2 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 2, }
dimensions: {natom: 3, nkpt: 6, mband: 7, nsppol: 1, nspinor: 1, nspden: 1, mpw: 472, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 9.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 4.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 0, iscf: 7, paral_kgb: 0, }
...
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 3.7796667 -3.7796667 0.0000000 G(1)= 0.1322868 -0.1322868 0.0000000
R(2)= 3.7796667 3.7796667 0.0000000 G(2)= 0.1322868 0.1322868 0.0000000
R(3)= 0.0000000 0.0000000 22.6780002 G(3)= 0.0000000 0.0000000 0.0440956
Unit cell volume ucvol= 6.4795039E+02 bohr^3
Angles (23,13,12)= 9.00000000E+01 9.00000000E+01 9.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 12 12 54
ecut(hartree)= 6.000 => boxcut(ratio)= 2.03597
--------------------------------------------------------------------------------
_setup2: Arith. and geom. avg. npw (full set) are 457.750 457.648
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 6, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -6.9580753614557 -6.958E+00 4.958E-03 2.464E+02 7.064E-03 7.064E-03
ETOT 2 -7.0341165674131 -7.604E-02 1.241E-04 6.103E+00 1.212E-02 5.054E-03
ETOT 3 -7.0343551418444 -2.386E-04 4.564E-05 9.219E+00 5.986E-04 4.455E-03
ETOT 4 -7.0354857046293 -1.131E-03 1.902E-05 3.254E+00 4.037E-04 4.859E-03
ETOT 5 -7.0358265157435 -3.408E-04 1.808E-05 8.770E-01 2.181E-04 5.077E-03
ETOT 6 -7.0359094075961 -8.289E-05 1.415E-05 5.270E-02 2.113E-04 5.288E-03
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.33581490E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.33581490E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -4.21318102E-05 sigma(2 1)= 0.00000000E+00
scprqt: WARNING -
nstep= 6 was not enough SCF cycles to converge;
maximum force difference= 2.113E-04 exceeds toldff= 5.000E-05
--- !ResultsGS
iteration_state: {dtset: 2, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -8.289E-05, res2: 5.270E-02, residm: 1.415E-05, diffor: 2.113E-04, }
etotal : -7.03590941E+00
entropy : 0.00000000E+00
fermie : 6.27372079E-02
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.33581490E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.33581490E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -4.21318102E-05, ]
pressure_GPa: -2.2069E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6667E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.3333E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -5.28812435E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, 5.28812435E-03, ]
force_length_stats: {min: 0.00000000E+00, max: 5.28812435E-03, mean: 3.52541623E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.88543893
2 2.00000 0.91777441
3 2.00000 0.88543893
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-5.00000000000000E-01 5.00000000000000E-01 1.66666666666667E-01
0.00000000000000E+00 0.00000000000000E+00 3.33333333333333E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 5.28812E-03 2.49285E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -5.28812434798399E-03
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 5.28812434798399E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 1.19924084840352E-01
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -1.19924084840352E-01
Total energy (etotal) [Ha]= -7.03590940759609E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 6, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -7.0358630852715 -7.036E+00 6.859E-07 3.270E-01 1.491E-04 5.437E-03
ETOT 2 -7.0359678613206 -1.048E-04 2.071E-07 3.410E-03 2.178E-04 5.219E-03
ETOT 3 -7.0359683925808 -5.313E-07 1.912E-08 6.831E-03 2.867E-05 5.248E-03
ETOT 4 -7.0359693024456 -9.099E-07 1.101E-08 2.085E-03 8.747E-06 5.239E-03
At SCF step 4, forces are converged :
for the second time, max diff in force= 8.747E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.33836136E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.33836136E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -3.55680628E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -9.099E-07, res2: 2.085E-03, residm: 1.101E-08, diffor: 8.747E-06, }
etotal : -7.03596930E+00
entropy : 0.00000000E+00
fermie : 6.33280603E-02
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.33836136E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.33836136E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -3.55680628E-05, ]
pressure_GPa: -2.2762E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -2.3318E-04, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6667E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.3357E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -5.23933843E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, 5.23933843E-03, ]
force_length_stats: {min: 0.00000000E+00, max: 5.23933843E-03, mean: 3.49289229E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.88539155
2 2.00000 0.91655200
3 2.00000 0.88539155
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -5.28812434798399E-03
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.56462151294799E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -2.33183010376676E-04
-5.00000000000000E-01 5.00000000000000E-01 1.66666666666667E-01
0.00000000000000E+00 0.00000000000000E+00 3.33566516343710E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 5.23934E-03 2.46985E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -5.23933843244170E-03
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 5.23933843244170E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 1.18817717839595E-01
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -1.18817717839595E-01
Total energy (etotal) [Ha]= -7.03596930244561E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-5.98948E-05
Relative =-8.51270E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 6, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -7.0339056586242 -7.034E+00 2.675E-03 1.625E+00 1.386E-02 8.623E-03
ETOT 2 -7.0345346665223 -6.290E-04 8.901E-04 2.022E-01 9.239E-04 7.699E-03
ETOT 3 -7.0345488205705 -1.415E-05 2.070E-04 1.155E-01 1.012E-04 7.598E-03
ETOT 4 -7.0345513861822 -2.566E-06 1.723E-04 8.995E-02 2.739E-05 7.571E-03
ETOT 5 -7.0345598897518 -8.504E-06 1.332E-05 3.197E-02 2.729E-06 7.568E-03
At SCF step 5, forces are converged :
for the second time, max diff in force= 2.729E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.09741232E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.09741232E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 1.18764415E-04 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -8.504E-06, res2: 3.197E-02, residm: 1.332E-05, diffor: 2.729E-06, }
etotal : -7.03455989E+00
entropy : 0.00000000E+00
fermie : 6.91464451E-02
cartesian_stress_tensor: # hartree/bohr^3
- [ 2.09741232E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 2.09741232E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 1.18764415E-04, ]
pressure_GPa: -5.2786E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -2.5276E-02, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6667E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.5861E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, 7.56815246E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, -7.56815246E-03, ]
force_length_stats: {min: 0.00000000E+00, max: 7.56815246E-03, mean: 5.04543498E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.87563943
2 2.00000 0.86261628
3 2.00000 0.87563943
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -5.73203532390411E-01
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 8.13253692099040E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -2.52757530734496E-02
-5.00000000000000E-01 5.00000000000000E-01 1.66666666666667E-01
0.00000000000000E+00 0.00000000000000E+00 3.58609086406782E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 7.56815E-03 3.56766E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 7.56815246313278E-03
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -7.56815246313278E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 -0.00000000000000E+00 -1.71630562813725E-01
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 0.00000000000000E+00 1.71630562813725E-01
Total energy (etotal) [Ha]= -7.03455988975180E+00
Difference of energy with previous step (new-old):
Absolute (Ha)= 1.40941E-03
Relative = 2.00335E-04
--- Iteration: ( 4/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 4, icycle: 1, }
solver: {iscf: 7, nstep: 6, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -7.0367632848339 -7.037E+00 4.293E-03 1.101E+00 8.302E-03 7.334E-04
ETOT 2 -7.0371290431813 -3.658E-04 2.240E-05 2.251E-02 7.524E-04 1.904E-05
ETOT 3 -7.0371346390102 -5.596E-06 7.117E-06 2.810E-02 4.452E-05 6.356E-05
ETOT 4 -7.0371360761421 -1.437E-06 6.434E-06 1.683E-02 2.204E-05 8.560E-05
At SCF step 4, forces are converged :
for the second time, max diff in force= 2.204E-05 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.76890134E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.76890134E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.90061747E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 4, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -1.437E-06, res2: 1.683E-02, residm: 6.434E-06, diffor: 2.204E-05, }
etotal : -7.03713608E+00
entropy : 0.00000000E+00
fermie : 6.62023388E-02
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.76890134E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.76890134E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 2.90061747E-05, ]
pressure_GPa: -3.7540E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -1.0478E-02, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6667E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.4381E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, 8.56031455E-05, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, -8.56031455E-05, ]
force_length_stats: {min: 0.00000000E+00, max: 8.56031455E-05, mean: 5.70687637E-05, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.85240131
2 2.00000 0.88949445
3 2.00000 0.85240131
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -2.37613179114227E-01
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.79694656771422E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -1.04776954483211E-02
-5.00000000000000E-01 5.00000000000000E-01 1.66666666666667E-01
0.00000000000000E+00 0.00000000000000E+00 3.43811028781654E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 8.56031E-05 4.03537E-05 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 8.56031455221931E-05
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -8.56031455221931E-05
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 -0.00000000000000E+00 -1.94130814834530E-03
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 0.00000000000000E+00 1.94130814834530E-03
Total energy (etotal) [Ha]= -7.03713607614213E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.57619E-03
Relative =-3.66152E-04
At Broyd/MD step 4, gradients are converged :
max grad (force/stress) = 8.5603E-05 < tolmxf= 5.0000E-04 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 16.484E-08; max= 64.337E-07
reduced coordinates (array xred) for 3 atoms
0.000000000000 0.000000000000 -0.010477695448
-0.500000000000 0.500000000000 0.166666666667
0.000000000000 0.000000000000 0.343811028782
rms dE/dt= 9.1514E-04; max dE/dt= 1.9413E-03; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 -0.001941308148
2 0.000000000000 0.000000000000 0.000000000000
3 0.000000000000 0.000000000000 0.001941308148
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 -0.12573947931216
2 0.00000000000000 2.00011347807574 2.00011347807574
3 0.00000000000000 0.00000000000000 4.12596643546363
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 0.00008560314552
2 -0.00000000000000 -0.00000000000000 -0.00000000000000
3 -0.00000000000000 -0.00000000000000 -0.00008560314552
frms,max,avg= 4.0353710E-05 8.5603146E-05 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 0.00440189072821
2 -0.00000000000000 -0.00000000000000 -0.00000000000000
3 -0.00000000000000 -0.00000000000000 -0.00440189072821
frms,max,avg= 2.0750712E-03 4.4018907E-03 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.559333388600 7.559333388600 7.559333388600 bohr
= 4.000226956151 4.000226956151 4.000226956151 angstroms
prteigrs : about to open file tbase4_6o_DS2_EIG
Fermi (or HOMO) energy (hartree) = 0.06620 Average Vxc (hartree)= -0.24487
Eigenvalues (hartree) for nkpt= 6 k points:
kpt# 1, nband= 7, wtk= 0.12500, kpt= -0.1250 0.0000 0.0000 (reduced coord)
-0.29581 -0.21866 -0.10461 0.00665 0.13547 0.18751 0.25525
occupation numbers for kpt# 1
2.00000 2.00000 2.00000 2.11575 0.00522 0.00012 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 2, itime: 4, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 2.36492041171901E+00
hartree : 6.63668374514960E+00
xc : -3.21026900039128E+00
Ewald energy : 3.52370928679226E-01
psp_core : 2.39348290026351E-02
local_psp : -1.44848946454702E+01
non_local_psp : 1.28084264464458E+00
internal : -7.03641108666646E+00
'-kT*entropy' : -7.24989475666252E-04
total_energy : -7.03713607614213E+00
total_energy_eV : -1.91490227833431E+02
band_energy : -6.65291282333152E-01
...
rms coord change= 4.9392E-03 atom, delta coord (reduced):
1 0.000000000000 0.000000000000 -0.010477695448
2 0.000000000000 0.000000000000 0.000000000000
3 0.000000000000 0.000000000000 0.010477695448
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.76890134E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.76890134E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.90061747E-05 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= -3.7540E+00 GPa]
- sigma(1 1)= 5.20428742E+00 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= 5.20428742E+00 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= 8.53391122E-01 sigma(2 1)= 0.00000000E+00
== END DATASET(S) ==============================================================
================================================================================
-outvars: echo values of variables after computation --------
acell 7.5593333886E+00 7.5593333886E+00 7.5593333886E+00 Bohr
amu 2.69815390E+01
ecut 6.00000000E+00 Hartree
etotal1 -7.0358659542E+00
etotal2 -7.0371360761E+00
fcart1 -0.0000000000E+00 -0.0000000000E+00 -2.9623755058E-04
-0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
-0.0000000000E+00 -0.0000000000E+00 2.9623755058E-04
fcart2 -0.0000000000E+00 -0.0000000000E+00 8.5603145522E-05
-0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
-0.0000000000E+00 -0.0000000000E+00 -8.5603145522E-05
- fftalg 512
ionmov 2
ixc -1012
jdtset 1 2
kpt -1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
-2.50000000E-01 1.25000000E-01 0.00000000E+00
5.00000000E-01 1.25000000E-01 0.00000000E+00
-3.75000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 3.75000000E-01 0.00000000E+00
kptrlatt 4 4 0 -4 4 0 0 0 1
kptrlen 3.02373336E+01
P mkmem 6
natom 3
nband 7
ndtset 2
ngfft1 12 12 45
ngfft2 12 12 54
nkpt 6
nstep 6
nsym 16
ntime 10
ntypat 1
occ1 2.000000 2.000000 2.000000 2.135255 0.003075 0.000197
0.000000
2.000000 2.000000 2.000676 2.016677 2.120277 0.100948
0.005238
2.000000 2.000000 2.000128 1.478645 0.839587 0.004872
0.001206
2.000000 2.000000 2.000263 2.000639 1.716166 0.587648
0.003087
2.000000 2.000001 2.037822 2.141840 1.012771 0.004591
0.002679
2.000424 2.000706 2.134034 2.122163 0.000535 0.001242
0.002464
occ2 2.000000 2.000000 2.000000 2.115753 0.005217 0.000125
0.000000
2.000000 2.000000 2.001094 2.017356 2.110698 0.181484
0.005238
2.000000 2.000000 2.000135 1.686744 0.679380 0.004665
0.000481
2.000000 2.000000 2.000308 2.000825 1.684466 0.609457
0.003911
2.000000 2.000001 2.049922 2.141779 0.941021 0.003163
0.002043
2.000730 2.001124 2.129884 2.108350 0.000120 0.002510
0.002679
occopt 4
optforces 1
rprim1 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 2.5000000000E+00
rprim2 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 3.0000000000E+00
shiftk 5.00000000E-01 5.00000000E-01 0.00000000E+00
spgroup 123
strten1 1.9248733879E-04 1.9248733879E-04 1.6535996526E-05
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten2 1.7689013407E-04 1.7689013407E-04 2.9006174654E-05
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
-1 0 0 0 1 0 0 0 -1 1 0 0 0 -1 0 0 0 1
-1 0 0 0 -1 0 0 0 1 1 0 0 0 1 0 0 0 -1
1 0 0 0 -1 0 0 0 -1 -1 0 0 0 1 0 0 0 1
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 -1 0 1 0 0 0 0 -1 0 1 0 -1 0 0 0 0 1
0 -1 0 -1 0 0 0 0 1 0 1 0 1 0 0 0 0 -1
0 1 0 -1 0 0 0 0 -1 0 -1 0 1 0 0 0 0 1
tnons1 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
tnons2 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.3333333
0.0000000 0.0000000 0.3333333 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.3333333
0.0000000 0.0000000 0.3333333 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.3333333
0.0000000 0.0000000 0.3333333 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.3333333
0.0000000 0.0000000 0.3333333 0.0000000 0.0000000 0.0000000
toldff 5.00000000E-05
tolmxf 5.00000000E-04
tsmear 4.00000000E-02 Hartree
typat 1 1 1
wtk 0.12500 0.12500 0.25000 0.12500 0.25000 0.12500
xangst1 0.0000000000E+00 0.0000000000E+00 -9.3734098997E-02
0.0000000000E+00 2.0001134781E+00 2.0001134781E+00
0.0000000000E+00 0.0000000000E+00 4.0939610551E+00
xangst2 0.0000000000E+00 0.0000000000E+00 -1.2573947931E-01
0.0000000000E+00 2.0001134781E+00 2.0001134781E+00
0.0000000000E+00 0.0000000000E+00 4.1259664355E+00
xcart1 0.0000000000E+00 0.0000000000E+00 -1.7713177576E-01
0.0000000000E+00 3.7796666943E+00 3.7796666943E+00
0.0000000000E+00 0.0000000000E+00 7.7364651644E+00
xcart2 0.0000000000E+00 0.0000000000E+00 -2.3761317911E-01
0.0000000000E+00 3.7796666943E+00 3.7796666943E+00
0.0000000000E+00 0.0000000000E+00 7.7969465677E+00
xred1 0.0000000000E+00 0.0000000000E+00 -9.3728780916E-03
-5.0000000000E-01 5.0000000000E-01 2.0000000000E-01
0.0000000000E+00 0.0000000000E+00 4.0937287809E-01
xred2 0.0000000000E+00 0.0000000000E+00 -1.0477695448E-02
-5.0000000000E-01 5.0000000000E-01 1.6666666667E-01
0.0000000000E+00 0.0000000000E+00 3.4381102878E-01
znucl 13.00000
================================================================================
- Timing analysis has been suppressed with timopt=0
================================================================================
Suggested references for the acknowledgment of ABINIT usage.
The users of ABINIT have little formal obligations with respect to the ABINIT group
(those specified in the GNU General Public License, http://www.gnu.org/copyleft/gpl.txt).
However, it is common practice in the scientific literature,
to acknowledge the efforts of people that have made the research possible.
In this spirit, please find below suggested citations of work written by ABINIT developers,
corresponding to implementations inside of ABINIT that you have used in the present run.
Note also that it will be of great value to readers of publications presenting these results,
to read papers enabling them to understand the theoretical formalism and details
of the ABINIT implementation.
For information on why they are suggested, see also https://docs.abinit.org/theory/acknowledgments.
-
- [1] Libxc: A library of exchange and correlation functionals for density functional theory.
- M.A.L. Marques, M.J.T. Oliveira, T. Burnus, Computer Physics Communications 183, 2227 (2012).
- Comment: to be cited when LibXC is used (negative value of ixc)
- Strong suggestion to cite this paper.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#marques2012
-
- [2] The Abinit project: Impact, environment and recent developments.
- Computer Phys. Comm. 248, 107042 (2020).
- X.Gonze, B. Amadon, G. Antonius, F.Arnardi, L.Baguet, J.-M.Beuken,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, N.Brouwer, F.Bruneval,
- G.Brunin, T.Cavignac, J.-B. Charraud, Wei Chen, M.Cote, S.Cottenier,
- J.Denier, G.Geneste, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, Xu He, N.Helbig, N.Holzwarth, Y.Jia, F.Jollet,
- W.Lafargue-Dit-Hauret, K.Lejaeghere, M.A.L.Marques, A.Martin, C.Martins,
- H.P.C. Miranda, F.Naccarato, K. Persson, G.Petretto, V.Planes, Y.Pouillon,
- S.Prokhorenko, F.Ricci, G.-M.Rignanese, A.H.Romero, M.M.Schmitt, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, G.Zerah and J.W.Zwanzig
- Comment: the fifth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT20.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2020
-
- [3] Optimized norm-conserving Vanderbilt pseudopotentials.
- D.R. Hamann, Phys. Rev. B 88, 085117 (2013).
- Comment: Some pseudopotential generated using the ONCVPSP code were used.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#hamann2013
-
- [4] ABINIT: Overview, and focus on selected capabilities
- J. Chem. Phys. 152, 124102 (2020).
- A. Romero, D.C. Allan, B. Amadon, G. Antonius, T. Applencourt, L.Baguet,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, F.Bruneval,
- G.Brunin, D.Caliste, M.Cote,
- J.Denier, C. Dreyer, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, F.Jollet, G. Jomard,
- A.Martin,
- H.P.C. Miranda, F.Naccarato, G.Petretto, N.A. Pike, V.Planes,
- S.Prokhorenko, T. Rangel, F.Ricci, G.-M.Rignanese, M.Royo, M.Stengel, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, J.Wiktor, J.W.Zwanziger, and X.Gonze.
- Comment: a global overview of ABINIT, with focus on selected capabilities .
- Note that a version of this paper, that is not formatted for J. Chem. Phys
- is available at https://www.abinit.org/sites/default/files/ABINIT20_JPC.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#romero2020
-
- [5] Recent developments in the ABINIT software package.
- Computer Phys. Comm. 205, 106 (2016).
- X.Gonze, F.Jollet, F.Abreu Araujo, D.Adams, B.Amadon, T.Applencourt,
- C.Audouze, J.-M.Beuken, J.Bieder, A.Bokhanchuk, E.Bousquet, F.Bruneval
- D.Caliste, M.Cote, F.Dahm, F.Da Pieve, M.Delaveau, M.Di Gennaro,
- B.Dorado, C.Espejo, G.Geneste, L.Genovese, A.Gerossier, M.Giantomassi,
- Y.Gillet, D.R.Hamann, L.He, G.Jomard, J.Laflamme Janssen, S.Le Roux,
- A.Levitt, A.Lherbier, F.Liu, I.Lukacevic, A.Martin, C.Martins,
- M.J.T.Oliveira, S.Ponce, Y.Pouillon, T.Rangel, G.-M.Rignanese,
- A.H.Romero, B.Rousseau, O.Rubel, A.A.Shukri, M.Stankovski, M.Torrent,
- M.J.Van Setten, B.Van Troeye, M.J.Verstraete, D.Waroquier, J.Wiktor,
- B.Xu, A.Zhou, J.W.Zwanziger.
- Comment: the fourth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT16.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2016
-
- Proc. 0 individual time (sec): cpu= 6.9 wall= 7.0
================================================================================
Calculation completed.
.Delivered 2 WARNINGs and 3 COMMENTs to log file.
+Overall time at end (sec) : cpu= 6.9 wall= 7.0
The run is on the order of of few seconds on a modern PC.
In the Broyden step 0 of the first dataset, you will notice the WARNING:
scprqt: WARNING -
nstep= 6 was not enough SCF cycles to converge;
maximum force difference= 6.859E-05 exceeds toldff= 5.000E-05
The input variable nstep was intentionally set to the rather low value of 6, to warn you about possible convergence difficulties. The SCF convergence might indeed get more and more difficult with cell size. This is because the default preconditioner (see the notice of the input variable dielng) is not very good for the metal+vacuum case. For the interpretation of the present run, this is not critical, as the convergence criterion was close of being fulfilled, but one should keep this in mind, as you will see.
For the 2 vacuum layer case, one has the non-relaxed total energy:
ETOT 6 -7.0350152828531
giving the unrelaxed surface energy 0.0193 Ha = 0.525 eV; and for the relaxed case:
etotal1 -7.0358659542E+00
(this one is converged to the required level) giving the relaxed surface energy 0.0189 Ha = 0.514 eV
Note that the difference between unrelaxed and relaxed case is a bit larger than in the case of one vacuum layer. This is because there was some interaction between slabs of different supercells.
For the 3 vacuum layer case, the self-consistency is slightly more difficult than with 2 vacuum layers: the Broyden step 0 is not sufficiently converged (one might set nstep to a larger value, but the best is to change the preconditioner, as described below)… However, for the Broyden steps number 2 and beyond, because one takes advantage of the previous wavefunctions, a sufficient convergence is reached. The total energy, in the relaxed case, is:
etotal2 -7.0371360761E+00
giving the relaxed surface energy 0.0182 Ha = 0.495 eV.
There is a rather small 0.019 eV difference with the 2 vacuum layer case.
For the next run, we will keep the 2 vacuum layer case, and we know that the precision of the coming calculation cannot be better than 0.019 eV. One might investigate the 4 vacuum layer case, but this is not worth, in the present tutorial.
Surface energy: increasing the number of aluminum layers¶
One should now increase the number of aluminum layers, while keeping 2 vacuum layers. We will consider 4 and 5 aluminum layers. This is rather straightforward to set up, but will also change the preconditioner. One could use an effective dielectric constant of about 3 or 5, with a rather small mixing coefficient, on the order of 0.2. However, there is also another possibility, using an estimation of the dielectric matrix governed by iprcel=45. For comparison with the previous treatment of SCF, one can recompute the result with 3 aluminum layers.
The input file tbase4_7.abi is an example, while
# Crystalline aluminum : computation of the total energy # # Determination of the surface energy of aluminum : # convergence with respect to the number of vacuum layers. ndtset 3 #Definition of the unit cell acell 3*7.5593333886E+00 # Lattice parameters of bulk aluminum rprim1 0.5 -0.5 0.0 0.5 0.5 0.0 0.0 0.0 2.5 rprim2 0.5 -0.5 0.0 0.5 0.5 0.0 0.0 0.0 3.0 rprim3 0.5 -0.5 0.0 0.5 0.5 0.0 0.0 0.0 3.5 natom1 3 # Three atoms per cell: three aluminum layers and some vacuum natom2 4 # Four atoms per cell: four aluminum layers and some vacuum natom3 5 # Five atoms per cell: five aluminum layers and some vacuum #Definition of the atom types ntypat 1 # There is only one type of atom znucl 13 # The keyword "znucl" refers to the atomic number of the # possible type(s) of atom. The pseudopotential(s) # mentioned in the "files" file must correspond # to the type(s) of atom. Here, the only type is Aluminum pp_dirpath "$ABI_PSPDIR" # This is the path to the directory were # pseudopotentials for tests are stored pseudos "Psdj_nc_sr_04_pw_std_psp8/Al.psp8" # Name and location of the pseudopotential #Definition of the atoms typat 1 1 1 1 1 # All possible atoms are type 1. xcart 3*0.0 # Triplet giving the CARTESIAN coordinates of atom 1. 0.0 2*3.7796666943 # Triplet giving the CARTESIAN coordinates of atom 2. 2*0.0 7.5593333886 # Triplet giving the CARTESIAN coordinates of atom 3. 0.0 3.7796666943 11.3390000829 # Triplet giving the CARTESIAN coordinates of atom 4. 2*0.0 15.1186667772 # Triplet giving the CARTESIAN coordinates of atom 5. chksymtnons 0 # Some of the non-symmorphic vectors are unconvential, but this is harmless in a ground-state calculation. #Definition of the planewave basis set ecut 6.0 # Maximal kinetic energy cut-off, in Hartree #SCF preconditioner iprcel 45 #Definition of the k-point grids ngkpt 4 4 1 nshiftk 2 shiftk 0.5 0.0 0.0 0.0 0.5 0.0 #Definition of the SCF procedure nstep 10 # Maximal number of SCF cycles toldff 5.0d-5 #Definition of occupation numbers occopt 4 tsmear 0.04 #The relaxation geoopt "bfgs" tolmxf 5.0d-4 ntime 10 ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% [files] #%% files_to_test = #%% tbase4_7.abo, tolnlines= 41, tolabs=1.001e-02, tolrel=0.03, fld_options=-easy #%% [paral_info] #%% max_nprocs = 4 #%% [extra_info] #%% authors = Unknown #%% keywords = #%% description = #%% Crystalline aluminum : computation of the total energy #%% Determination of the surface energy of aluminum : #%% convergence with respect to the number of vacuum layers. #%%<END TEST_INFO>
tbase4_7.abo is a reference output file.
.Version 10.5.8.2 of ABINIT, released Oct 2025.
.(MPI version, prepared for a x86_64_linux_gnu13.2 computer)
.Copyright (C) 1998-2026 ABINIT group .
ABINIT comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Sat 20 Dec 2025.
- ( at 17h06 )
- input file -> /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/TestBot_MPI1/tutorial_tbase4_7/tbase4_7.abi
- output file -> tbase4_7.abo
- root for input files -> tbase4_7i
- root for output files -> tbase4_7o
DATASET 1 : space group P4/m m m (#123); Bravais tP (primitive tetrag.)
================================================================================
Values of the parameters that define the memory need for DATASET 1.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 45 mpssoang = 3 mqgrid = 3001
natom = 3 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 16 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 7 mffmem = 1 mkmem = 6
mpw = 388 nfft = 6480 nkpt = 6
For the susceptibility and dielectric matrices, or tddft :
mgfft = 30 nbnd_in_blk= 4 nfft = 1920 npw = 85
================================================================================
P This job should need less than 4.112 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.251 Mbytes ; DEN or POT disk file : 0.051 Mbytes.
================================================================================
DATASET 2 : space group P4/n m m (#129); Bravais tP (primitive tetrag.)
================================================================================
Values of the parameters that define the memory need for DATASET 2.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 54 mpssoang = 3 mqgrid = 3001
natom = 4 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 16 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 8 mffmem = 1 mkmem = 6
mpw = 472 nfft = 7776 nkpt = 6
For the susceptibility and dielectric matrices, or tddft :
mgfft = 36 nbnd_in_blk= 4 nfft = 2304 npw = 103
================================================================================
P This job should need less than 4.767 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.348 Mbytes ; DEN or POT disk file : 0.061 Mbytes.
================================================================================
DATASET 3 : space group P4/m m m (#123); Bravais tP (primitive tetrag.)
================================================================================
Values of the parameters that define the memory need for DATASET 3.
intxc = 0 ionmov = 2 iscf = 7 lmnmax = 6
lnmax = 6 mgfft = 60 mpssoang = 3 mqgrid = 3001
natom = 5 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 16 n1xccc = 2501 ntypat = 1
occopt = 4 xclevel = 1
- mband = 11 mffmem = 1 mkmem = 6
mpw = 544 nfft = 8640 nkpt = 6
For the susceptibility and dielectric matrices, or tddft :
mgfft = 40 nbnd_in_blk= 6 nfft = 2560 npw = 121
================================================================================
P This job should need less than 5.413 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 0.550 Mbytes ; DEN or POT disk file : 0.068 Mbytes.
================================================================================
--------------------------------------------------------------------------------
------------- Echo of variables that govern the present computation ------------
--------------------------------------------------------------------------------
-
- outvars: echo of selected default values
- iomode0 = 0 , fftalg0 =512 , wfoptalg0 = 0
-
- outvars: echo of global parameters not present in the input file
- max_nthreads = 0
-
-outvars: echo values of preprocessed input variables --------
acell 7.5593333886E+00 7.5593333886E+00 7.5593333886E+00 Bohr
amu 2.69815390E+01
chksymtnons 0
ecut 6.00000000E+00 Hartree
- fftalg 512
ionmov 2
iprcel 45
ixc -1012
jdtset 1 2 3
kpt -1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
-2.50000000E-01 1.25000000E-01 0.00000000E+00
5.00000000E-01 1.25000000E-01 0.00000000E+00
-3.75000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 3.75000000E-01 0.00000000E+00
kptrlatt 4 4 0 -4 4 0 0 0 1
kptrlen 3.02373336E+01
P mkmem 6
natom1 3
natom2 4
natom3 5
nband1 7
nband2 8
nband3 11
ndtset 3
ngfft1 12 12 45
ngfft2 12 12 54
ngfft3 12 12 60
nkpt 6
nstep 10
nsym 16
ntime 10
ntypat 1
occ1 2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
2.000000 2.000000 2.000000 2.000000 1.000000 0.000000
0.000000
occ2 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
0.000000 0.000000
2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
0.000000 0.000000
2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
0.000000 0.000000
2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
0.000000 0.000000
2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
0.000000 0.000000
2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
0.000000 0.000000
occ3 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 1.000000 0.000000 0.000000 0.000000
2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 1.000000 0.000000 0.000000 0.000000
2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 1.000000 0.000000 0.000000 0.000000
2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 1.000000 0.000000 0.000000 0.000000
2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 1.000000 0.000000 0.000000 0.000000
2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 1.000000 0.000000 0.000000 0.000000
occopt 4
optforces 1
rprim1 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 2.5000000000E+00
rprim2 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 3.0000000000E+00
rprim3 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 3.5000000000E+00
shiftk 5.00000000E-01 5.00000000E-01 0.00000000E+00
spgroup1 123
spgroup2 129
spgroup3 123
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
-1 0 0 0 1 0 0 0 -1 1 0 0 0 -1 0 0 0 1
-1 0 0 0 -1 0 0 0 1 1 0 0 0 1 0 0 0 -1
1 0 0 0 -1 0 0 0 -1 -1 0 0 0 1 0 0 0 1
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 -1 0 1 0 0 0 0 -1 0 1 0 -1 0 0 0 0 1
0 -1 0 -1 0 0 0 0 1 0 1 0 1 0 0 0 0 -1
0 1 0 -1 0 0 0 0 -1 0 -1 0 1 0 0 0 0 1
tnons1 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
tnons2 0.0000000 0.0000000 0.0000000 0.5000000 0.5000000 0.5000000
0.5000000 0.5000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.5000000 0.5000000 0.5000000
0.5000000 0.5000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.5000000 0.5000000 0.5000000
0.5000000 0.5000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.5000000 0.5000000 0.5000000
0.5000000 0.5000000 0.5000000 0.0000000 0.0000000 0.0000000
tnons3 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 -0.4285714
0.0000000 0.0000000 -0.4285714 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 -0.4285714
0.0000000 0.0000000 -0.4285714 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 -0.4285714
0.0000000 0.0000000 -0.4285714 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 -0.4285714
0.0000000 0.0000000 -0.4285714 0.0000000 0.0000000 0.0000000
toldff 5.00000000E-05
tolmxf 5.00000000E-04
tsmear 4.00000000E-02 Hartree
typat1 1 1 1
typat2 1 1 1 1
typat3 1 1 1 1 1
wtk 0.12500 0.12500 0.25000 0.12500 0.25000 0.12500
xangst1 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 2.0001134781E+00 2.0001134781E+00
0.0000000000E+00 0.0000000000E+00 4.0002269562E+00
xangst2 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 2.0001134781E+00 2.0001134781E+00
0.0000000000E+00 0.0000000000E+00 4.0002269562E+00
0.0000000000E+00 2.0001134781E+00 6.0003404342E+00
xangst3 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 2.0001134781E+00 2.0001134781E+00
0.0000000000E+00 0.0000000000E+00 4.0002269562E+00
0.0000000000E+00 2.0001134781E+00 6.0003404342E+00
0.0000000000E+00 0.0000000000E+00 8.0004539123E+00
xcart1 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 3.7796666943E+00 3.7796666943E+00
0.0000000000E+00 0.0000000000E+00 7.5593333886E+00
xcart2 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 3.7796666943E+00 3.7796666943E+00
0.0000000000E+00 0.0000000000E+00 7.5593333886E+00
0.0000000000E+00 3.7796666943E+00 1.1339000083E+01
xcart3 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 3.7796666943E+00 3.7796666943E+00
0.0000000000E+00 0.0000000000E+00 7.5593333886E+00
0.0000000000E+00 3.7796666943E+00 1.1339000083E+01
0.0000000000E+00 0.0000000000E+00 1.5118666777E+01
xred1 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
-5.0000000000E-01 5.0000000000E-01 2.0000000000E-01
0.0000000000E+00 0.0000000000E+00 4.0000000000E-01
xred2 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
-5.0000000000E-01 5.0000000000E-01 1.6666666667E-01
0.0000000000E+00 0.0000000000E+00 3.3333333333E-01
-5.0000000000E-01 5.0000000000E-01 5.0000000000E-01
xred3 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
-5.0000000000E-01 5.0000000000E-01 1.4285714286E-01
0.0000000000E+00 0.0000000000E+00 2.8571428571E-01
-5.0000000000E-01 5.0000000000E-01 4.2857142857E-01
0.0000000000E+00 0.0000000000E+00 5.7142857143E-01
znucl 13.00000
================================================================================
chkinp: Checking input parameters for consistency, jdtset= 1.
chkinp: Checking input parameters for consistency, jdtset= 2.
chkinp: Checking input parameters for consistency, jdtset= 3.
================================================================================
== DATASET 1 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 1, }
dimensions: {natom: 3, nkpt: 6, mband: 7, nsppol: 1, nspinor: 1, nspden: 1, mpw: 388, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 9.00000000E+00, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 4.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 0, iscf: 7, paral_kgb: 0, }
...
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 3.7796667 -3.7796667 0.0000000 G(1)= 0.1322868 -0.1322868 0.0000000
R(2)= 3.7796667 3.7796667 0.0000000 G(2)= 0.1322868 0.1322868 0.0000000
R(3)= 0.0000000 0.0000000 18.8983335 G(3)= 0.0000000 0.0000000 0.0529147
Unit cell volume ucvol= 5.3995866E+02 bohr^3
Angles (23,13,12)= 9.00000000E+01 9.00000000E+01 9.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 12 12 45
ecut(hartree)= 6.000 => boxcut(ratio)= 2.03597
--- Pseudopotential description ------------------------------------------------
- pspini: atom type 1 psp file is /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/Pspdir/Psdj_nc_sr_04_pw_std_psp8/Al.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/trunk__codata2022/tests/Pspdir/Psdj_nc_sr_04_pw_std_psp8/Al.psp8
- Al ONCVPSP-3.3.0 r_core= 1.76802 1.76802 1.70587
- 13.00000 3.00000 171102 znucl, zion, pspdat
8 -1012 2 4 600 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
5.99000000000000 5.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2 2
extension_switch 1
pspatm : epsatm= 0.57439192
--- l ekb(1:nproj) -->
0 5.725870 0.726131
1 6.190420 0.914022
2 -4.229503 -0.925599
pspatm: atomic psp has been read and splines computed
1.55085818E+01 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
_setup2: Arith. and geom. avg. npw (full set) are 379.250 379.160
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -6.9582155601713 -6.958E+00 1.194E-02 1.964E+02 6.735E-03 6.735E-03
ETOT 2 -7.0324432447447 -7.423E-02 6.889E-03 7.528E+00 1.191E-02 5.171E-03
ETOT 3 -7.0350032510037 -2.560E-03 7.601E-05 7.125E-03 6.866E-04 4.484E-03
ETOT 4 -7.0350151234939 -1.187E-05 3.123E-05 4.615E-04 9.439E-05 4.579E-03
ETOT 5 -7.0350152998026 -1.763E-07 1.206E-06 4.389E-06 1.011E-05 4.569E-03
ETOT 6 -7.0350153035194 -3.717E-09 4.453E-07 9.984E-09 1.309E-06 4.567E-03
At SCF step 6, forces are converged :
for the second time, max diff in force= 1.309E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.50570219E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.50570219E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -3.72412265E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 1, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 18.8983335, ]
lattice_lengths: [ 5.34526, 5.34526, 18.89833, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 5.3995866E+02
convergence: {deltae: -3.717E-09, res2: 9.984E-09, residm: 4.453E-07, diffor: 1.309E-06, }
etotal : -7.03501530E+00
entropy : 0.00000000E+00
fermie : 1.11354439E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.50570219E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.50570219E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -3.72412265E-05, ]
pressure_GPa: -2.5881E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
- [ -5.0000E-01, 5.0000E-01, 2.0000E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 4.0000E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -4.56732363E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -2.89120579E-19, ]
- [ -0.00000000E+00, -0.00000000E+00, 4.56732363E-03, ]
force_length_stats: {min: 2.89120579E-19, max: 4.56732363E-03, mean: 3.04488242E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.88605825
2 2.00000 0.91706811
3 2.00000 0.88605825
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-5.00000000000000E-01 5.00000000000000E-01 2.00000000000000E-01
0.00000000000000E+00 0.00000000000000E+00 4.00000000000000E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 4.56732E-03 2.15306E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -4.56732363193391E-03
-0.00000000000000E+00 -0.00000000000000E+00 -2.89120579329468E-19
-0.00000000000000E+00 -0.00000000000000E+00 4.56732363193391E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 8.63148050685496E-02
-0.00000000000000E+00 0.00000000000000E+00 5.46389712164155E-18
-0.00000000000000E+00 -0.00000000000000E+00 -8.63148050685496E-02
Total energy (etotal) [Ha]= -7.03501530351936E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -7.0350557538698 -7.035E+00 2.885E-08 1.350E-03 1.145E-04 4.453E-03
ETOT 2 -7.0350564775471 -7.237E-07 5.171E-10 5.355E-05 3.852E-06 4.449E-03
ETOT 3 -7.0350564958283 -1.828E-08 2.008E-10 4.193E-07 3.122E-06 4.452E-03
At SCF step 3, forces are converged :
for the second time, max diff in force= 3.122E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.51782929E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.51782929E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -3.57858080E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 1, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 18.8983335, ]
lattice_lengths: [ 5.34526, 5.34526, 18.89833, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 5.3995866E+02
convergence: {deltae: -1.828E-08, res2: 4.193E-07, residm: 2.008E-10, diffor: 3.122E-06, }
etotal : -7.03505650E+00
entropy : 0.00000000E+00
fermie : 1.11409459E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.51782929E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.51782929E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -3.57858080E-05, ]
pressure_GPa: -2.6261E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -2.4168E-04, Al]
- [ -5.0000E-01, 5.0000E-01, 2.0000E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 4.0024E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -4.45208485E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, 2.89120579E-19, ]
- [ -0.00000000E+00, -0.00000000E+00, 4.45208485E-03, ]
force_length_stats: {min: 2.89120579E-19, max: 4.45208485E-03, mean: 2.96805657E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.88611695
2 2.00000 0.91646125
3 2.00000 0.88611695
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -4.56732363193391E-03
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.56390071223194E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -2.41678645306040E-04
-5.00000000000000E-01 5.00000000000000E-01 2.00000000000000E-01
0.00000000000000E+00 0.00000000000000E+00 4.00241678645306E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 4.45208E-03 2.09873E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -4.45208484979823E-03
-0.00000000000000E+00 -0.00000000000000E+00 2.89120579329468E-19
-0.00000000000000E+00 -0.00000000000000E+00 4.45208484979823E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 8.41369841349000E-02
-0.00000000000000E+00 -0.00000000000000E+00 -5.46389712164155E-18
-0.00000000000000E+00 -0.00000000000000E+00 -8.41369841348999E-02
Total energy (etotal) [Ha]= -7.03505649582832E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-4.11923E-05
Relative =-5.85531E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 1, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -7.0353691480727 -7.035E+00 5.398E-06 9.811E-01 4.527E-03 7.502E-05
ETOT 2 -7.0358486702824 -4.795E-04 1.455E-07 5.579E-02 2.306E-04 1.556E-04
ETOT 3 -7.0358679831122 -1.931E-05 2.350E-07 9.472E-04 6.309E-05 2.187E-04
ETOT 4 -7.0358683746156 -3.915E-07 4.119E-09 5.860E-07 7.829E-06 2.108E-04
ETOT 5 -7.0358683756898 -1.074E-09 1.582E-09 1.741E-08 1.105E-06 2.097E-04
At SCF step 5, forces are converged :
for the second time, max diff in force= 1.105E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.93094798E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.93094798E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 1.95979885E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 1, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 18.8983335, ]
lattice_lengths: [ 5.34526, 5.34526, 18.89833, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 5.3995866E+02
convergence: {deltae: -1.074E-09, res2: 1.741E-08, residm: 1.582E-09, diffor: 1.105E-06, }
etotal : -7.03586838E+00
entropy : 0.00000000E+00
fermie : 1.13484678E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.93094798E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.93094798E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 1.95979885E-05, ]
pressure_GPa: -3.9796E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -9.5786E-03, Al]
- [ -5.0000E-01, 5.0000E-01, 2.0000E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 4.0958E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -2.09724873E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, 2.09724873E-04, ]
force_length_stats: {min: 0.00000000E+00, max: 2.09724873E-04, mean: 1.39816582E-04, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.85788358
2 2.00000 0.89527301
3 2.00000 0.85788358
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -1.81019312875598E-01
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.74035270147561E+00
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -9.57858602445488E-03
-5.00000000000000E-01 5.00000000000000E-01 2.00000000000000E-01
0.00000000000000E+00 0.00000000000000E+00 4.09578586024456E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 2.09725E-04 9.88653E-05 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -2.09724873461449E-04
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 2.09724873461449E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 3.96345059594261E-03
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -3.96345059594261E-03
Total energy (etotal) [Ha]= -7.03586837568975E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-8.11880E-04
Relative =-1.15398E-04
At Broyd/MD step 3, gradients are converged :
max grad (force/stress) = 2.0972E-04 < tolmxf= 5.0000E-04 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 41.824E-12; max= 15.818E-10
reduced coordinates (array xred) for 3 atoms
0.000000000000 0.000000000000 -0.009578586024
-0.500000000000 0.500000000000 0.200000000000
0.000000000000 0.000000000000 0.409578586024
rms dE/dt= 1.8684E-03; max dE/dt= 3.9635E-03; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.003963450596
2 0.000000000000 0.000000000000 0.000000000000
3 0.000000000000 0.000000000000 -0.003963450596
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 -0.09579129504210
2 0.00000000000000 2.00011347807574 2.00011347807574
3 0.00000000000000 0.00000000000000 4.09601825119358
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00020972487346
2 -0.00000000000000 -0.00000000000000 -0.00000000000000
3 -0.00000000000000 -0.00000000000000 0.00020972487346
frms,max,avg= 9.8865253E-05 2.0972487E-04 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.01078448660191
2 -0.00000000000000 -0.00000000000000 -0.00000000000000
3 -0.00000000000000 -0.00000000000000 0.01078448660191
frms,max,avg= 5.0838557E-03 1.0784487E-02 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.559333388600 7.559333388600 7.559333388600 bohr
= 4.000226956151 4.000226956151 4.000226956151 angstroms
prteigrs : about to open file tbase4_7o_DS1_EIG
Fermi (or HOMO) energy (hartree) = 0.11348 Average Vxc (hartree)= -0.28264
Eigenvalues (hartree) for nkpt= 6 k points:
kpt# 1, nband= 7, wtk= 0.12500, kpt= -0.1250 0.0000 0.0000 (reduced coord)
-0.25207 -0.17269 -0.05788 0.05950 0.17231 0.23124 0.30463
occupation numbers for kpt# 1
2.00000 2.00000 2.00000 2.13484 0.00291 0.00019 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 1, itime: 3, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 2.38516616029097E+00
hartree : 3.30165030927567E+00
xc : -3.21940413329354E+00
Ewald energy : -3.41056914214704E+00
psp_core : 2.87217948031621E-02
local_psp : -7.40352958307986E+00
non_local_psp : 1.28340075073743E+00
internal : -7.03456384341321E+00
'-kT*entropy' : -1.30453227654255E-03
total_energy : -7.03586837568975E+00
total_energy_eV : -1.91455731946777E+02
band_energy : -2.53039287603314E-01
...
rms coord change= 4.5154E-03 atom, delta coord (reduced):
1 0.000000000000 0.000000000000 -0.009578586024
2 0.000000000000 0.000000000000 0.000000000000
3 0.000000000000 0.000000000000 0.009578586024
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.93094798E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.93094798E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 1.95979885E-05 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= -3.9796E+00 GPa]
- sigma(1 1)= 5.68104508E+00 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= 5.68104508E+00 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= 5.76592730E-01 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 2 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 2, }
dimensions: {natom: 4, nkpt: 6, mband: 8, nsppol: 1, nspinor: 1, nspden: 1, mpw: 472, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 1.20000000E+01, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 4.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 0, iscf: 7, paral_kgb: 0, }
...
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 3.7796667 -3.7796667 0.0000000 G(1)= 0.1322868 -0.1322868 0.0000000
R(2)= 3.7796667 3.7796667 0.0000000 G(2)= 0.1322868 0.1322868 0.0000000
R(3)= 0.0000000 0.0000000 22.6780002 G(3)= 0.0000000 0.0000000 0.0440956
Unit cell volume ucvol= 6.4795039E+02 bohr^3
Angles (23,13,12)= 9.00000000E+01 9.00000000E+01 9.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 12 12 54
ecut(hartree)= 6.000 => boxcut(ratio)= 2.03597
2.75708122E+01 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
_setup2: Arith. and geom. avg. npw (full set) are 457.750 457.648
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -9.2825257493648 -9.283E+00 3.557E-03 4.317E+02 4.968E-03 4.968E-03
ETOT 2 -9.3897228909704 -1.072E-01 1.499E-03 2.376E+01 1.155E-02 6.578E-03
ETOT 3 -9.3957971983268 -6.074E-03 5.815E-05 6.339E-02 1.450E-03 6.348E-03
ETOT 4 -9.3958282562896 -3.106E-05 5.466E-05 6.577E-03 1.157E-04 6.422E-03
ETOT 5 -9.3958298984572 -1.642E-06 2.481E-06 4.731E-05 1.349E-05 6.409E-03
ETOT 6 -9.3958299123969 -1.394E-08 4.356E-06 1.726E-06 1.432E-06 6.408E-03
At SCF step 6, forces are converged :
for the second time, max diff in force= 1.432E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.63715234E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.63715234E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -4.11982087E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -1.394E-08, res2: 1.726E-06, residm: 4.356E-06, diffor: 1.432E-06, }
etotal : -9.39582991E+00
entropy : 0.00000000E+00
fermie : 1.42418293E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.63715234E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.63715234E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -4.11982087E-05, ]
pressure_GPa: -2.8071E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6667E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.3333E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 5.0000E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -6.40751242E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, 7.20382022E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -7.20382022E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, 6.40751242E-03, ]
force_length_stats: {min: 7.20382022E-04, max: 6.40751242E-03, mean: 3.56394722E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.87903101
2 2.00000 0.92300217
3 2.00000 0.92300217
4 2.00000 0.87903101
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
0.00000000000000E+00 3.77966669430000E+00 1.13390000829000E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-5.00000000000000E-01 5.00000000000000E-01 1.66666666666667E-01
0.00000000000000E+00 0.00000000000000E+00 3.33333333333333E-01
-5.00000000000000E-01 5.00000000000000E-01 5.00000000000000E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 6.40751E-03 2.63234E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -6.40751242363954E-03
-0.00000000000000E+00 -0.00000000000000E+00 7.20382022454053E-04
-0.00000000000000E+00 -0.00000000000000E+00 -7.20382022454053E-04
-0.00000000000000E+00 -0.00000000000000E+00 6.40751242363954E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 1.45309567805663E-01
-0.00000000000000E+00 -0.00000000000000E+00 -1.63368236246524E-02
-0.00000000000000E+00 0.00000000000000E+00 1.63368236246524E-02
-0.00000000000000E+00 -0.00000000000000E+00 -1.45309567805663E-01
Total energy (etotal) [Ha]= -9.39582991239687E+00
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -9.3959087900973 -9.396E+00 3.581E-07 7.090E-03 3.075E-04 6.161E-03
ETOT 2 -9.3959112552582 -2.465E-06 1.933E-08 3.031E-04 2.904E-05 6.166E-03
ETOT 3 -9.3959113269808 -7.172E-08 9.234E-10 5.244E-06 4.665E-06 6.169E-03
At SCF step 3, forces are converged :
for the second time, max diff in force= 4.665E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.64672148E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.64672148E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -3.85624304E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -7.172E-08, res2: 5.244E-06, residm: 9.234E-10, diffor: 4.665E-06, }
etotal : -9.39591133E+00
entropy : 0.00000000E+00
fermie : 1.42433806E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.64672148E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.64672148E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -3.85624304E-05, ]
pressure_GPa: -2.8517E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -2.8254E-04, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6670E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.3330E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 5.0028E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -6.16947052E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, 4.37307545E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -4.37307545E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, 6.16947052E-03, ]
force_length_stats: {min: 4.37307545E-04, max: 6.16947052E-03, mean: 3.30338903E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.87912809
2 2.00000 0.92260746
3 2.00000 0.92260746
4 2.00000 0.87912809
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -6.40751242363954E-03
0.00000000000000E+00 3.77966669430000E+00 3.78038707632245E+00
0.00000000000000E+00 0.00000000000000E+00 7.55861300657755E+00
0.00000000000000E+00 3.77966669430000E+00 1.13454075953236E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -2.82543098015429E-04
-5.00000000000000E-01 5.00000000000000E-01 1.66698432343410E-01
0.00000000000000E+00 0.00000000000000E+00 3.33301567656590E-01
-5.00000000000000E-01 5.00000000000000E-01 5.00282543098016E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 6.16947E-03 2.52500E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -6.16947052268146E-03
-0.00000000000000E+00 -0.00000000000000E+00 4.37307545287509E-04
-0.00000000000000E+00 -0.00000000000000E+00 -4.37307545287509E-04
-0.00000000000000E+00 -0.00000000000000E+00 6.16947052268146E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 1.39911253536268E-01
-0.00000000000000E+00 -0.00000000000000E+00 -9.91726058453572E-03
-0.00000000000000E+00 0.00000000000000E+00 9.91726058453573E-03
-0.00000000000000E+00 -0.00000000000000E+00 -1.39911253536268E-01
Total energy (etotal) [Ha]= -9.39591132698082E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-8.14146E-05
Relative =-8.66493E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -9.3959533494700 -9.396E+00 4.084E-06 2.890E+00 6.660E-03 6.222E-03
ETOT 2 -9.3968922957443 -9.389E-04 4.368E-07 1.521E-01 6.732E-04 5.549E-03
ETOT 3 -9.3969286454643 -3.635E-05 3.261E-07 2.737E-03 9.690E-05 5.646E-03
ETOT 4 -9.3969294253484 -7.799E-07 1.932E-08 4.375E-06 6.634E-06 5.640E-03
ETOT 5 -9.3969294285494 -3.201E-09 1.228E-08 1.651E-07 1.334E-06 5.642E-03
At SCF step 5, forces are converged :
for the second time, max diff in force= 1.334E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.82523954E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.82523954E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.41699843E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -3.201E-09, res2: 1.651E-07, residm: 1.228E-08, diffor: 1.334E-06, }
etotal : -9.39692943E+00
entropy : 0.00000000E+00
fermie : 1.42630463E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.82523954E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.82523954E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 2.41699843E-05, ]
pressure_GPa: -3.8171E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -7.0783E-03, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6719E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.3281E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 5.0708E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -9.18303608E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -5.64171074E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, 5.64171074E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, 9.18303608E-04, ]
force_length_stats: {min: 9.18303608E-04, max: 5.64171074E-03, mean: 3.28000717E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.87136391
2 2.00000 0.91356588
3 2.00000 0.91356588
4 2.00000 0.87136391
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -1.60521982202716E-01
0.00000000000000E+00 3.77966669430000E+00 3.79155122716906E+00
0.00000000000000E+00 0.00000000000000E+00 7.54744885573095E+00
0.00000000000000E+00 3.77966669430000E+00 1.14995220651027E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -7.07831294775250E-03
-5.00000000000000E-01 5.00000000000000E-01 1.67190722261612E-01
0.00000000000000E+00 0.00000000000000E+00 3.32809277738389E-01
-5.00000000000000E-01 5.00000000000000E-01 5.07078312947753E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 5.64171E-03 2.33353E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -9.18303608290987E-04
-0.00000000000000E+00 -0.00000000000000E+00 -5.64171073736609E-03
-0.00000000000000E+00 -0.00000000000000E+00 5.64171073736609E-03
-0.00000000000000E+00 -0.00000000000000E+00 9.18303608290987E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 2.08252893810777E-02
-0.00000000000000E+00 0.00000000000000E+00 1.27942717037384E-01
-0.00000000000000E+00 -0.00000000000000E+00 -1.27942717037384E-01
-0.00000000000000E+00 -0.00000000000000E+00 -2.08252893810777E-02
Total energy (etotal) [Ha]= -9.39692942854940E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.01810E-03
Relative =-1.08350E-04
--- Iteration: ( 4/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 4, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -9.3969791100740 -9.397E+00 2.388E-07 8.716E-02 4.723E-04 5.961E-03
ETOT 2 -9.3970084081592 -2.930E-05 8.538E-09 4.074E-03 9.841E-05 5.863E-03
ETOT 3 -9.3970093885672 -9.804E-07 1.790E-08 7.203E-05 1.456E-05 5.877E-03
ETOT 4 -9.3970094088478 -2.028E-08 3.119E-10 8.979E-08 1.243E-06 5.876E-03
At SCF step 4, forces are converged :
for the second time, max diff in force= 1.243E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.84957969E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.84957969E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.17570056E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 4, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -2.028E-08, res2: 8.979E-08, residm: 3.119E-10, diffor: 1.243E-06, }
etotal : -9.39700941E+00
entropy : 0.00000000E+00
fermie : 1.42718910E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.84957969E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.84957969E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.17570056E-05, ]
pressure_GPa: -3.9392E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -7.9687E-03, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6699E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.3301E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 5.0797E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -4.64804732E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -5.87619920E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, 5.87619920E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, 4.64804732E-04, ]
force_length_stats: {min: 4.64804732E-04, max: 5.87619920E-03, mean: 3.17050197E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.85176413
2 2.00000 0.91210169
3 2.00000 0.91210169
4 2.00000 0.85176413
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -1.80713265021438E-01
0.00000000000000E+00 3.77966669430000E+00 3.78702930750449E+00
0.00000000000000E+00 0.00000000000000E+00 7.55197077539553E+00
0.00000000000000E+00 3.77966669430000E+00 1.15197133479215E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -7.96865965694658E-03
-5.00000000000000E-01 5.00000000000000E-01 1.66991325505659E-01
0.00000000000000E+00 0.00000000000000E+00 3.33008674494342E-01
-5.00000000000000E-01 5.00000000000000E-01 5.07968659656947E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 5.87620E-03 2.40644E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -4.64804731667080E-04
-0.00000000000000E+00 -0.00000000000000E+00 -5.87619919973871E-03
-0.00000000000000E+00 -0.00000000000000E+00 5.87619919973871E-03
-0.00000000000000E+00 -0.00000000000000E+00 4.64804731667080E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 1.05408417818107E-02
-0.00000000000000E+00 0.00000000000000E+00 1.33260446425948E-01
-0.00000000000000E+00 -0.00000000000000E+00 -1.33260446425948E-01
-0.00000000000000E+00 -0.00000000000000E+00 -1.05408417818107E-02
Total energy (etotal) [Ha]= -9.39700940884782E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-7.99803E-05
Relative =-8.51129E-06
--- Iteration: ( 5/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 5, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -9.3971051889169 -9.397E+00 1.161E-05 8.484E-01 1.764E-03 5.650E-03
ETOT 2 -9.3974059075897 -3.007E-04 2.859E-07 4.428E-02 2.717E-04 5.378E-03
ETOT 3 -9.3974163348027 -1.043E-05 2.921E-07 1.851E-03 4.463E-05 5.419E-03
ETOT 4 -9.3974168229424 -4.881E-07 7.493E-08 1.239E-06 6.952E-06 5.412E-03
At SCF step 4, forces are converged :
for the second time, max diff in force= 6.952E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.98299227E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.98299227E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 6.75401530E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 5, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -4.881E-07, res2: 1.239E-06, residm: 7.493E-08, diffor: 6.952E-06, }
etotal : -9.39741682E+00
entropy : 0.00000000E+00
fermie : 1.43405878E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.98299227E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.98299227E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 6.75401530E-05, ]
pressure_GPa: -4.5518E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -1.2428E-02, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6513E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.3487E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 5.1243E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, 1.12046564E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -5.41231173E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, 5.41231173E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -1.12046564E-03, ]
force_length_stats: {min: 1.12046564E-03, max: 5.41231173E-03, mean: 3.26638868E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.87692889
2 2.00000 0.90369196
3 2.00000 0.90369196
4 2.00000 0.87692889
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -2.81835135660784E-01
0.00000000000000E+00 3.77966669430000E+00 3.74480142929663E+00
0.00000000000000E+00 0.00000000000000E+00 7.59419865360340E+00
0.00000000000000E+00 3.77966669430000E+00 1.16208352185608E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -1.24276891083990E-02
-5.00000000000000E-01 5.00000000000000E-01 1.65129261924252E-01
0.00000000000000E+00 0.00000000000000E+00 3.34870738075749E-01
-5.00000000000000E-01 5.00000000000000E-01 5.12427689108401E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 5.41231E-03 2.25642E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 1.12046563684474E-03
-0.00000000000000E+00 -0.00000000000000E+00 -5.41231172838481E-03
-0.00000000000000E+00 -0.00000000000000E+00 5.41231172838481E-03
-0.00000000000000E+00 -0.00000000000000E+00 -1.12046563684474E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 -0.00000000000000E+00 -2.54099198981383E-02
-0.00000000000000E+00 0.00000000000000E+00 1.22740406273672E-01
-0.00000000000000E+00 -0.00000000000000E+00 -1.22740406273672E-01
-0.00000000000000E+00 0.00000000000000E+00 2.54099198981383E-02
Total energy (etotal) [Ha]= -9.39741682294245E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-4.07414E-04
Relative =-4.33548E-05
--- Iteration: ( 6/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 6, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -9.3976377378656 -9.398E+00 5.743E-06 8.771E-02 1.771E-03 3.642E-03
ETOT 2 -9.3976783632359 -4.063E-05 3.337E-07 4.055E-03 1.025E-04 3.613E-03
ETOT 3 -9.3976792984545 -9.352E-07 3.139E-07 2.744E-04 1.567E-05 3.624E-03
ETOT 4 -9.3976793700367 -7.158E-08 4.490E-08 7.207E-07 2.672E-06 3.621E-03
At SCF step 4, forces are converged :
for the second time, max diff in force= 2.672E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.07312983E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.07312983E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 8.61353374E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 6, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -7.158E-08, res2: 7.207E-07, residm: 4.490E-08, diffor: 2.672E-06, }
etotal : -9.39767937E+00
entropy : 0.00000000E+00
fermie : 1.44062371E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 2.07312983E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 2.07312983E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 8.61353374E-05, ]
pressure_GPa: -4.9110E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -1.5042E-02, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6311E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.3689E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 5.1504E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, 1.39770731E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -3.62096821E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, 3.62096821E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -1.39770731E-03, ]
force_length_stats: {min: 1.39770731E-03, max: 3.62096821E-03, mean: 2.50933776E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.87516277
2 2.00000 0.90794518
3 2.00000 0.90794518
4 2.00000 0.87516277
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -3.41130133901987E-01
0.00000000000000E+00 3.77966669430000E+00 3.69899666649580E+00
0.00000000000000E+00 0.00000000000000E+00 7.64000341640424E+00
0.00000000000000E+00 3.77966669430000E+00 1.16801302168020E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -1.50423375698019E-02
-5.00000000000000E-01 5.00000000000000E-01 1.63109473474391E-01
0.00000000000000E+00 0.00000000000000E+00 3.36890526525611E-01
-5.00000000000000E-01 5.00000000000000E-01 5.15042337569804E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 3.62097E-03 1.58456E-03 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 1.39770730961334E-03
-0.00000000000000E+00 -0.00000000000000E+00 -3.62096821309254E-03
-0.00000000000000E+00 -0.00000000000000E+00 3.62096821309254E-03
-0.00000000000000E+00 -0.00000000000000E+00 -1.39770730961334E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 -0.00000000000000E+00 -3.16972065991512E-02
-0.00000000000000E+00 0.00000000000000E+00 8.21163177368692E-02
-0.00000000000000E+00 -0.00000000000000E+00 -8.21163177368692E-02
-0.00000000000000E+00 0.00000000000000E+00 3.16972065991512E-02
Total energy (etotal) [Ha]= -9.39767937003672E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.62547E-04
Relative =-2.79378E-05
--- Iteration: ( 7/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 7, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -9.3978007463577 -9.398E+00 1.811E-06 1.358E-01 2.509E-03 1.112E-03
ETOT 2 -9.3978340450253 -3.330E-05 2.090E-07 7.087E-03 1.058E-04 1.211E-03
ETOT 3 -9.3978358361553 -1.791E-06 6.566E-08 5.352E-05 1.403E-05 1.197E-03
ETOT 4 -9.3978358526536 -1.650E-08 1.408E-08 3.732E-07 1.092E-06 1.197E-03
At SCF step 4, forces are converged :
for the second time, max diff in force= 1.092E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.12465629E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.12465629E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 9.03666387E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 7, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -1.650E-08, res2: 3.732E-07, residm: 1.408E-08, diffor: 1.092E-06, }
etotal : -9.39783585E+00
entropy : 0.00000000E+00
fermie : 1.44588411E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 2.12465629E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 2.12465629E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 9.03666387E-05, ]
pressure_GPa: -5.0535E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -1.6033E-02, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6122E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.3878E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 5.1603E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, 7.63720010E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -1.19702993E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, 1.19702993E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -7.63720010E-04, ]
force_length_stats: {min: 7.63720010E-04, max: 1.19702993E-03, mean: 9.80374971E-04, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.86700892
2 2.00000 0.89670944
3 2.00000 0.89670944
4 2.00000 0.86700892
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -3.63599113911943E-01
0.00000000000000E+00 3.77966669430000E+00 3.65615105961678E+00
0.00000000000000E+00 0.00000000000000E+00 7.68284902328323E+00
0.00000000000000E+00 3.77966669430000E+00 1.17025991968119E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -1.60331207008401E-02
-5.00000000000000E-01 5.00000000000000E-01 1.61220170777250E-01
0.00000000000000E+00 0.00000000000000E+00 3.38779829222750E-01
-5.00000000000000E-01 5.00000000000000E-01 5.16033120700840E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 1.19703E-03 5.79676E-04 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 7.63720010260221E-04
-0.00000000000000E+00 -0.00000000000000E+00 -1.19702993260316E-03
-0.00000000000000E+00 -0.00000000000000E+00 1.19702993260316E-03
-0.00000000000000E+00 -0.00000000000000E+00 -7.63720010260221E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 -0.00000000000000E+00 -1.73196425193061E-02
-0.00000000000000E+00 0.00000000000000E+00 2.71462450100419E-02
-0.00000000000000E+00 -0.00000000000000E+00 -2.71462450100419E-02
-0.00000000000000E+00 0.00000000000000E+00 1.73196425193061E-02
Total energy (etotal) [Ha]= -9.39783585265361E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.56483E-04
Relative =-1.66511E-05
--- Iteration: ( 8/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 2, itime: 8, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -9.3978525573898 -9.398E+00 3.742E-08 2.729E-02 1.125E-03 1.661E-04
ETOT 2 -9.3978592099104 -6.653E-06 4.762E-09 1.698E-03 4.942E-05 1.609E-04
ETOT 3 -9.3978596458036 -4.359E-07 2.050E-09 1.624E-06 6.034E-06 1.670E-04
At SCF step 3, forces are converged :
for the second time, max diff in force= 6.034E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.12581146E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.12581146E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 8.55585965E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 2, itime: 8, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 22.6780002, ]
lattice_lengths: [ 5.34526, 5.34526, 22.67800, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 6.4795039E+02
convergence: {deltae: -4.359E-07, res2: 1.624E-06, residm: 2.050E-09, diffor: 6.034E-06, }
etotal : -9.39785965E+00
entropy : 0.00000000E+00
fermie : 1.44677754E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 2.12581146E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 2.12581146E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 8.55585965E-05, ]
pressure_GPa: -5.0086E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -1.5635E-02, Al]
- [ -5.0000E-01, 5.0000E-01, 1.6070E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 3.3930E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 5.1564E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, 1.66982526E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -1.16166518E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, 1.16166518E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -1.66982526E-04, ]
force_length_stats: {min: 1.16166518E-04, max: 1.66982526E-04, mean: 1.41574522E-04, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.86734644
2 2.00000 0.90666899
3 2.00000 0.90666899
4 2.00000 0.86734644
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -3.54577021511602E-01
0.00000000000000E+00 3.77966669430000E+00 3.64443146572107E+00
0.00000000000000E+00 0.00000000000000E+00 7.69456861717892E+00
0.00000000000000E+00 3.77966669430000E+00 1.16935771044116E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -1.56352861327838E-02
-5.00000000000000E-01 5.00000000000000E-01 1.60703388265122E-01
0.00000000000000E+00 0.00000000000000E+00 3.39296611734877E-01
-5.00000000000000E-01 5.00000000000000E-01 5.15635286132783E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 1.66983E-04 8.30440E-05 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 1.66982526479608E-04
-0.00000000000000E+00 -0.00000000000000E+00 -1.16166518494392E-04
-0.00000000000000E+00 -0.00000000000000E+00 1.16166518494392E-04
-0.00000000000000E+00 -0.00000000000000E+00 -1.66982526479608E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 -0.00000000000000E+00 -3.78682976319026E-03
-0.00000000000000E+00 0.00000000000000E+00 2.63442432567622E-03
-0.00000000000000E+00 -0.00000000000000E+00 -2.63442432567622E-03
-0.00000000000000E+00 0.00000000000000E+00 3.78682976319026E-03
Total energy (etotal) [Ha]= -9.39785964580358E+00
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.37931E-05
Relative =-2.53177E-06
At Broyd/MD step 8, gradients are converged :
max grad (force/stress) = 1.6698E-04 < tolmxf= 5.0000E-04 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 82.045E-11; max= 20.502E-10
reduced coordinates (array xred) for 4 atoms
0.000000000000 0.000000000000 -0.015635286133
-0.500000000000 0.500000000000 0.160703388265
0.000000000000 0.000000000000 0.339296611735
-0.500000000000 0.500000000000 0.515635286133
rms dE/dt= 1.8833E-03; max dE/dt= 3.7868E-03; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 -0.003786829763
2 0.000000000000 0.000000000000 0.002634424326
3 0.000000000000 0.000000000000 -0.002634424326
4 0.000000000000 0.000000000000 0.003786829763
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 -0.18763407916651
2 0.00000000000000 2.00011347807574 1.92855007704905
3 0.00000000000000 0.00000000000000 4.07179035717814
4 0.00000000000000 2.00011347807574 6.18797451339371
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 0.00016698252648
2 -0.00000000000000 -0.00000000000000 -0.00011616651849
3 -0.00000000000000 -0.00000000000000 0.00011616651849
4 -0.00000000000000 -0.00000000000000 -0.00016698252648
frms,max,avg= 8.3043989E-05 1.6698253E-04 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 0.00858658674982
2 -0.00000000000000 -0.00000000000000 -0.00597352255656
3 -0.00000000000000 -0.00000000000000 0.00597352255656
4 -0.00000000000000 -0.00000000000000 -0.00858658674982
frms,max,avg= 4.2702936E-03 8.5865867E-03 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.559333388600 7.559333388600 7.559333388600 bohr
= 4.000226956151 4.000226956151 4.000226956151 angstroms
prteigrs : about to open file tbase4_7o_DS2_EIG
Fermi (or HOMO) energy (hartree) = 0.14468 Average Vxc (hartree)= -0.30112
Eigenvalues (hartree) for nkpt= 6 k points:
kpt# 1, nband= 8, wtk= 0.12500, kpt= -0.1250 0.0000 0.0000 (reduced coord)
-0.23142 -0.18513 -0.11195 -0.01221 0.06737 0.20797 0.25290 0.25949
occupation numbers for kpt# 1
2.00000 2.00000 2.00000 2.00000 2.04429 0.00432 0.00055 0.00027
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 2, itime: 8, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 3.19425602202794E+00
hartree : 4.47031606188327E+00
xc : -4.31139210921522E+00
Ewald energy : -4.80194323558358E+00
psp_core : 4.25508071157957E-02
local_psp : -9.68828992122671E+00
non_local_psp : 1.69835752177656E+00
internal : -9.39614485322194E+00
'-kT*entropy' : -1.71479258163333E-03
total_energy : -9.39785964580358E+00
total_energy_eV : -2.55728788707479E+02
band_energy : -4.21605656986125E-02
...
rms coord change= 6.8316E-03 atom, delta coord (reduced):
1 0.000000000000 0.000000000000 -0.015635286133
2 0.000000000000 0.000000000000 -0.005963278402
3 0.000000000000 0.000000000000 0.005963278402
4 0.000000000000 0.000000000000 0.015635286133
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 2.12581146E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 2.12581146E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 8.55585965E-05 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= -5.0086E+00 GPa]
- sigma(1 1)= 6.25435325E+00 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= 6.25435325E+00 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= 2.51722082E+00 sigma(2 1)= 0.00000000E+00
================================================================================
== DATASET 3 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 3, }
dimensions: {natom: 5, nkpt: 6, mband: 11, nsppol: 1, nspinor: 1, nspden: 1, mpw: 544, }
cutoff_energies: {ecut: 6.0, pawecutdg: -1.0, }
electrons: {nelect: 1.50000000E+01, charge: 0.00000000E+00, occopt: 4.00000000E+00, tsmear: 4.00000000E-02, }
meta: {optdriver: 0, ionmov: 2, optcell: 0, iscf: 7, paral_kgb: 0, }
...
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 3.7796667 -3.7796667 0.0000000 G(1)= 0.1322868 -0.1322868 0.0000000
R(2)= 3.7796667 3.7796667 0.0000000 G(2)= 0.1322868 0.1322868 0.0000000
R(3)= 0.0000000 0.0000000 26.4576669 G(3)= 0.0000000 0.0000000 0.0377962
Unit cell volume ucvol= 7.5594212E+02 bohr^3
Angles (23,13,12)= 9.00000000E+01 9.00000000E+01 9.00000000E+01 degrees
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 12 12 60
ecut(hartree)= 6.000 => boxcut(ratio)= 2.03597
4.30793940E+01 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
_setup2: Arith. and geom. avg. npw (full set) are 531.750 531.680
================================================================================
=== [ionmov= 2] Broyden-Fletcher-Goldfarb-Shanno method (forces)
================================================================================
--- Iteration: ( 1/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 3, itime: 1, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -11.565348603830 -1.157E+01 8.366E-03 9.359E+02 1.830E-02 1.830E-02
ETOT 2 -11.743183678967 -1.778E-01 4.622E-04 6.144E+01 2.083E-02 2.540E-03
ETOT 3 -11.754728127644 -1.154E-02 2.112E-04 4.690E-02 1.730E-03 2.477E-03
ETOT 4 -11.754755115881 -2.699E-05 3.625E-05 3.189E-03 6.856E-05 2.532E-03
ETOT 5 -11.754755832143 -7.163E-07 2.132E-05 2.825E-05 1.594E-05 2.516E-03
ETOT 6 -11.754755842794 -1.065E-08 1.207E-05 3.931E-07 5.899E-07 2.515E-03
At SCF step 6, forces are converged :
for the second time, max diff in force= 5.899E-07 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42196301E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.42196301E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.30061222E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 3, itime: 1, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 26.4576669, ]
lattice_lengths: [ 5.34526, 5.34526, 26.45767, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 7.5594212E+02
convergence: {deltae: -1.065E-08, res2: 3.931E-07, residm: 1.207E-05, diffor: 5.899E-07, }
etotal : -1.17547558E+01
entropy : 0.00000000E+00
fermie : 1.67304831E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.42196301E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.42196301E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -2.30061222E-05, ]
pressure_GPa: -2.5634E+00
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Al]
- [ -5.0000E-01, 5.0000E-01, 1.4286E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 2.8571E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 4.2857E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 5.7143E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -2.51546087E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -8.89712223E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -8.67361738E-20, ]
- [ -0.00000000E+00, -0.00000000E+00, 8.89712223E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, 2.51546087E-03, ]
force_length_stats: {min: 8.67361738E-20, max: 2.51546087E-03, mean: 1.36206924E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.88993964
2 2.00000 0.89670165
3 2.00000 0.93735033
4 2.00000 0.92746518
5 2.00000 0.89173161
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
0.00000000000000E+00 3.77966669430000E+00 3.77966669430000E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
0.00000000000000E+00 3.77966669430000E+00 1.13390000829000E+01
0.00000000000000E+00 0.00000000000000E+00 1.51186667772000E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-5.00000000000000E-01 5.00000000000000E-01 1.42857142857143E-01
0.00000000000000E+00 0.00000000000000E+00 2.85714285714286E-01
-5.00000000000000E-01 5.00000000000000E-01 4.28571428571429E-01
0.00000000000000E+00 0.00000000000000E+00 5.71428571428572E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 2.51546E-03 9.74278E-04 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -2.51546087186417E-03
-0.00000000000000E+00 -0.00000000000000E+00 -8.89712223265378E-04
-0.00000000000000E+00 -0.00000000000000E+00 -8.67361737988404E-20
-0.00000000000000E+00 -0.00000000000000E+00 8.89712223265378E-04
-0.00000000000000E+00 -0.00000000000000E+00 2.51546087186417E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 6.65532257473988E-02
-0.00000000000000E+00 0.00000000000000E+00 2.35397096045143E-02
-0.00000000000000E+00 0.00000000000000E+00 2.29483679108945E-18
-0.00000000000000E+00 -0.00000000000000E+00 -2.35397096045143E-02
-0.00000000000000E+00 -0.00000000000000E+00 -6.65532257473988E-02
Total energy (etotal) [Ha]= -1.17547558427944E+01
--- Iteration: ( 2/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 3, itime: 2, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -11.754768877842 -1.175E+01 8.876E-06 4.518E-03 5.414E-05 2.461E-03
ETOT 2 -11.754769952913 -1.075E-06 7.009E-06 1.063E-04 1.752E-05 2.465E-03
ETOT 3 -11.754769974250 -2.134E-08 6.705E-06 4.898E-07 1.833E-06 2.466E-03
At SCF step 3, forces are converged :
for the second time, max diff in force= 1.833E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42565689E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.42565689E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= -2.22311045E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 3, itime: 2, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 26.4576669, ]
lattice_lengths: [ 5.34526, 5.34526, 26.45767, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 7.5594212E+02
convergence: {deltae: -2.134E-08, res2: 4.898E-07, residm: 6.705E-06, diffor: 1.833E-06, }
etotal : -1.17547700E+01
entropy : 0.00000000E+00
fermie : 1.67324029E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.42565689E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.42565689E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, -2.22311045E-05, ]
pressure_GPa: -2.5783E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -9.5075E-05, Al]
- [ -5.0000E-01, 5.0000E-01, 1.4282E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 2.8571E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 4.2861E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 5.7152E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -2.46606670E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -9.08712455E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, 9.08712455E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, 2.46606670E-03, ]
force_length_stats: {min: 0.00000000E+00, max: 2.46606670E-03, mean: 1.34991166E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.89005296
2 2.00000 0.89652911
3 2.00000 0.93721680
4 2.00000 0.92728682
5 2.00000 0.89183619
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -2.51546087186417E-03
0.00000000000000E+00 3.77966669430000E+00 3.77877698207673E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
0.00000000000000E+00 3.77966669430000E+00 1.13398897951233E+01
0.00000000000000E+00 0.00000000000000E+00 1.51211822380719E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -9.50749317831066E-05
-5.00000000000000E-01 5.00000000000000E-01 1.42823515091400E-01
0.00000000000000E+00 0.00000000000000E+00 2.85714285714286E-01
-5.00000000000000E-01 5.00000000000000E-01 4.28605056337171E-01
0.00000000000000E+00 0.00000000000000E+00 5.71523646360355E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 2.46607E-03 9.59670E-04 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 -2.46606669561309E-03
-0.00000000000000E+00 -0.00000000000000E+00 -9.08712455080114E-04
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 9.08712455080114E-04
-0.00000000000000E+00 -0.00000000000000E+00 2.46606669561309E-03
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 0.00000000000000E+00 6.52463710873188E-02
-0.00000000000000E+00 0.00000000000000E+00 2.40424114081332E-02
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -2.40424114081332E-02
-0.00000000000000E+00 -0.00000000000000E+00 -6.52463710873188E-02
Total energy (etotal) [Ha]= -1.17547699742496E+01
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.41315E-05
Relative =-1.20219E-06
--- Iteration: ( 3/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 3, itime: 3, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -11.752952578872 -1.175E+01 4.619E-05 9.807E+00 3.426E-03 2.670E-03
ETOT 2 -11.755197660014 -2.245E-03 1.405E-05 3.049E-01 7.828E-04 1.887E-03
ETOT 3 -11.755258298923 -6.064E-05 1.214E-05 2.475E-03 8.979E-05 1.977E-03
ETOT 4 -11.755258826242 -5.273E-07 1.003E-05 1.309E-06 6.658E-06 1.970E-03
ETOT 5 -11.755258827108 -8.655E-10 6.122E-06 7.391E-08 1.128E-07 1.970E-03
At SCF step 5, forces are converged :
for the second time, max diff in force= 1.128E-07 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.65078328E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.65078328E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 2.86310157E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 3, itime: 3, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 26.4576669, ]
lattice_lengths: [ 5.34526, 5.34526, 26.45767, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 7.5594212E+02
convergence: {deltae: -8.655E-10, res2: 7.391E-08, residm: 6.122E-06, diffor: 1.128E-07, }
etotal : -1.17552588E+01
entropy : 0.00000000E+00
fermie : 1.68348890E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.65078328E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.65078328E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 2.86310157E-05, ]
pressure_GPa: -3.5186E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -6.3461E-03, Al]
- [ -5.0000E-01, 5.0000E-01, 1.4052E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 2.8571E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 4.3091E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 5.7777E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, 5.15466070E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -1.97037246E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, 2.16840434E-20, ]
- [ -0.00000000E+00, -0.00000000E+00, 1.97037246E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -5.15466070E-04, ]
force_length_stats: {min: 2.16840434E-20, max: 1.97037246E-03, mean: 9.94335413E-04, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.88454091
2 2.00000 0.88586372
3 2.00000 0.92852354
4 2.00000 0.91644414
5 2.00000 0.89498784
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -1.67903521285795E-01
0.00000000000000E+00 3.77966669430000E+00 3.71786087545993E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
0.00000000000000E+00 3.77966669430000E+00 1.14008059017401E+01
0.00000000000000E+00 0.00000000000000E+00 1.52865702984858E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -6.34611971545401E-03
-5.00000000000000E-01 5.00000000000000E-01 1.40521116057543E-01
0.00000000000000E+00 0.00000000000000E+00 2.85714285714286E-01
-5.00000000000000E-01 5.00000000000000E-01 4.30907455371028E-01
0.00000000000000E+00 0.00000000000000E+00 5.77774691144026E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 1.97037E-03 7.43691E-04 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 5.15466069592048E-04
-0.00000000000000E+00 -0.00000000000000E+00 -1.97037246262458E-03
-0.00000000000000E+00 -0.00000000000000E+00 2.16840434497101E-20
-0.00000000000000E+00 -0.00000000000000E+00 1.97037246262458E-03
-0.00000000000000E+00 -0.00000000000000E+00 -5.15466069592048E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 -0.00000000000000E+00 -1.36380295469515E-02
-0.00000000000000E+00 0.00000000000000E+00 5.21314582064359E-02
-0.00000000000000E+00 -0.00000000000000E+00 -5.73709197772363E-19
-0.00000000000000E+00 -0.00000000000000E+00 -5.21314582064359E-02
-0.00000000000000E+00 0.00000000000000E+00 1.36380295469515E-02
Total energy (etotal) [Ha]= -1.17552588271078E+01
Difference of energy with previous step (new-old):
Absolute (Ha)=-4.88853E-04
Relative =-4.15868E-05
--- Iteration: ( 4/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 3, itime: 4, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -11.755235862460 -1.176E+01 4.154E-06 1.450E-01 2.104E-04 2.016E-03
ETOT 2 -11.755270287027 -3.442E-05 1.787E-06 3.978E-03 8.738E-05 1.929E-03
ETOT 3 -11.755271093045 -8.060E-07 1.793E-06 2.026E-05 1.042E-05 1.939E-03
ETOT 4 -11.755271097518 -4.473E-09 7.930E-07 1.625E-08 5.951E-07 1.939E-03
At SCF step 4, forces are converged :
for the second time, max diff in force= 5.951E-07 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.66959630E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.66959630E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.29051617E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 3, itime: 4, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 26.4576669, ]
lattice_lengths: [ 5.34526, 5.34526, 26.45767, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 7.5594212E+02
convergence: {deltae: -4.473E-09, res2: 1.625E-08, residm: 7.930E-07, diffor: 5.951E-07, }
etotal : -1.17552711E+01
entropy : 0.00000000E+00
fermie : 1.68430865E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.66959630E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.66959630E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.29051617E-05, ]
pressure_GPa: -3.5974E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -6.8893E-03, Al]
- [ -5.0000E-01, 5.0000E-01, 1.4023E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 2.8571E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 4.3119E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 5.7832E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, 6.98183326E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -1.93875843E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -2.16840434E-20, ]
- [ -0.00000000E+00, -0.00000000E+00, 1.93875843E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -6.98183326E-04, ]
force_length_stats: {min: 2.16840434E-20, max: 1.93875843E-03, mean: 1.05477670E-03, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.86398252
2 2.00000 0.89496202
3 2.00000 0.92751157
4 2.00000 0.91548291
5 2.00000 0.90307020
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -1.82274346485825E-01
0.00000000000000E+00 3.77966669430000E+00 3.71025806421101E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
0.00000000000000E+00 3.77966669430000E+00 1.14084087129890E+01
0.00000000000000E+00 0.00000000000000E+00 1.53009411236858E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -6.88928269637817E-03
-5.00000000000000E-01 5.00000000000000E-01 1.40233758472722E-01
0.00000000000000E+00 0.00000000000000E+00 2.85714285714286E-01
-5.00000000000000E-01 5.00000000000000E-01 4.31194812955849E-01
0.00000000000000E+00 0.00000000000000E+00 5.78317854124950E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 1.93876E-03 7.52440E-04 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 6.98183326075214E-04
-0.00000000000000E+00 -0.00000000000000E+00 -1.93875842753075E-03
-0.00000000000000E+00 -0.00000000000000E+00 -2.16840434497101E-20
-0.00000000000000E+00 -0.00000000000000E+00 1.93875842753075E-03
-0.00000000000000E+00 -0.00000000000000E+00 -6.98183326075214E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 -0.00000000000000E+00 -1.84723018485746E-02
-0.00000000000000E+00 0.00000000000000E+00 5.12950245978200E-02
-0.00000000000000E+00 0.00000000000000E+00 5.73709197772363E-19
-0.00000000000000E+00 -0.00000000000000E+00 -5.12950245978200E-02
-0.00000000000000E+00 0.00000000000000E+00 1.84723018485746E-02
Total energy (etotal) [Ha]= -1.17552710975177E+01
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.22704E-05
Relative =-1.04382E-06
--- Iteration: ( 5/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 3, itime: 5, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -11.755217806499 -1.176E+01 1.265E-06 3.019E-01 2.166E-04 1.750E-03
ETOT 2 -11.755294707156 -7.690E-05 3.546E-07 8.100E-03 1.305E-04 1.619E-03
ETOT 3 -11.755296416941 -1.710E-06 3.089E-07 5.292E-05 1.559E-05 1.635E-03
ETOT 4 -11.755296428418 -1.148E-08 1.444E-07 6.152E-08 1.093E-06 1.634E-03
At SCF step 4, forces are converged :
for the second time, max diff in force= 1.093E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.70053775E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.70053775E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 3.96220111E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 3, itime: 5, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 26.4576669, ]
lattice_lengths: [ 5.34526, 5.34526, 26.45767, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 7.5594212E+02
convergence: {deltae: -1.148E-08, res2: 6.152E-08, residm: 1.444E-07, diffor: 1.093E-06, }
etotal : -1.17552964E+01
entropy : 0.00000000E+00
fermie : 1.68565060E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.70053775E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.70053775E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 3.96220111E-05, ]
pressure_GPa: -3.7240E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -7.7515E-03, Al]
- [ -5.0000E-01, 5.0000E-01, 1.3959E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 2.8571E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 4.3184E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 5.7918E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, 8.56435995E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -1.63395897E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, 1.63395897E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -8.56435995E-04, ]
force_length_stats: {min: 0.00000000E+00, max: 1.63395897E-03, mean: 9.96157986E-04, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.85725551
2 2.00000 0.91438771
3 2.00000 0.92532974
4 2.00000 0.91387668
5 2.00000 0.88262315
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -2.05086477462047E-01
0.00000000000000E+00 3.77966669430000E+00 3.69322848044989E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
0.00000000000000E+00 3.77966669430000E+00 1.14254382967501E+01
0.00000000000000E+00 0.00000000000000E+00 1.53237532546620E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -7.75149519216797E-03
-5.00000000000000E-01 5.00000000000000E-01 1.39590104447930E-01
0.00000000000000E+00 0.00000000000000E+00 2.85714285714286E-01
-5.00000000000000E-01 5.00000000000000E-01 4.31838466980642E-01
0.00000000000000E+00 0.00000000000000E+00 5.79180066620740E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 1.63396E-03 6.73627E-04 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 8.56435995310626E-04
-0.00000000000000E+00 -0.00000000000000E+00 -1.63395896988634E-03
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 1.63395896988634E-03
-0.00000000000000E+00 -0.00000000000000E+00 -8.56435995310626E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 -0.00000000000000E+00 -2.26592982509267E-02
-0.00000000000000E+00 0.00000000000000E+00 4.32307420883250E-02
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -4.32307420883250E-02
-0.00000000000000E+00 0.00000000000000E+00 2.26592982509267E-02
Total energy (etotal) [Ha]= -1.17552964284177E+01
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.53309E-05
Relative =-2.15485E-06
--- Iteration: ( 6/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 3, itime: 6, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -11.755293647868 -1.176E+01 3.704E-07 8.472E-02 5.548E-04 1.079E-03
ETOT 2 -11.755323619412 -2.997E-05 1.272E-07 1.797E-03 7.889E-05 1.000E-03
ETOT 3 -11.755324098724 -4.793E-07 4.919E-08 9.295E-06 8.822E-06 1.009E-03
ETOT 4 -11.755324100910 -2.186E-09 4.480E-08 3.877E-08 6.923E-07 1.008E-03
At SCF step 4, forces are converged :
for the second time, max diff in force= 6.923E-07 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.72242574E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.72242574E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.38985416E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 3, itime: 6, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 26.4576669, ]
lattice_lengths: [ 5.34526, 5.34526, 26.45767, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 7.5594212E+02
convergence: {deltae: -2.186E-09, res2: 3.877E-08, residm: 4.480E-08, diffor: 6.923E-07, }
etotal : -1.17553241E+01
entropy : 0.00000000E+00
fermie : 1.68661024E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.72242574E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.72242574E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 4.38985416E-05, ]
pressure_GPa: -3.8089E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -8.3133E-03, Al]
- [ -5.0000E-01, 5.0000E-01, 1.3885E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 2.8571E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 4.3257E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 5.7974E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, 7.40934684E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -1.00836498E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -4.33680869E-20, ]
- [ -0.00000000E+00, -0.00000000E+00, 1.00836498E-03, ]
- [ -0.00000000E+00, -0.00000000E+00, -7.40934684E-04, ]
force_length_stats: {min: 4.33680869E-20, max: 1.00836498E-03, mean: 6.99719866E-04, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.85620752
2 2.00000 0.92441473
3 2.00000 0.92294792
4 2.00000 0.91324559
5 2.00000 0.90099347
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -2.19950662019566E-01
0.00000000000000E+00 3.77966669430000E+00 3.67375267755387E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
0.00000000000000E+00 3.77966669430000E+00 1.14449140996461E+01
0.00000000000000E+00 0.00000000000000E+00 1.53386174392196E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -8.31330529568605E-03
-5.00000000000000E-01 5.00000000000000E-01 1.38853992567808E-01
0.00000000000000E+00 0.00000000000000E+00 2.85714285714286E-01
-5.00000000000000E-01 5.00000000000000E-01 4.32574578860764E-01
0.00000000000000E+00 0.00000000000000E+00 5.79741876724258E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 1.00836E-03 4.56915E-04 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 7.40934683766509E-04
-0.00000000000000E+00 -0.00000000000000E+00 -1.00836498113285E-03
-0.00000000000000E+00 -0.00000000000000E+00 -4.33680868994202E-20
-0.00000000000000E+00 -0.00000000000000E+00 1.00836498113285E-03
-0.00000000000000E+00 -0.00000000000000E+00 -7.40934683766509E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 -0.00000000000000E+00 -1.96034030281878E-02
-0.00000000000000E+00 0.00000000000000E+00 2.66789847442039E-02
-0.00000000000000E+00 0.00000000000000E+00 1.14741839554473E-18
-0.00000000000000E+00 -0.00000000000000E+00 -2.66789847442039E-02
-0.00000000000000E+00 0.00000000000000E+00 1.96034030281878E-02
Total energy (etotal) [Ha]= -1.17553241009100E+01
Difference of energy with previous step (new-old):
Absolute (Ha)=-2.76725E-05
Relative =-2.35404E-06
--- Iteration: ( 7/10) Internal Cycle: (1/1)
--------------------------------------------------------------------------------
---SELF-CONSISTENT-FIELD CONVERGENCE--------------------------------------------
--- !BeginCycle
iteration_state: {dtset: 3, itime: 7, icycle: 1, }
solver: {iscf: 7, nstep: 10, nline: 4, wfoptalg: 0, }
tolerances: {toldff: 5.00E-05, }
...
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor
ETOT 1 -11.755333311062 -1.176E+01 4.446E-09 2.531E-02 7.324E-04 3.874E-04
ETOT 2 -11.755342938420 -9.627E-06 9.653E-09 9.018E-04 4.680E-05 3.406E-04
ETOT 3 -11.755343136450 -1.980E-07 3.687E-09 8.329E-07 5.330E-06 3.460E-04
At SCF step 3, forces are converged :
for the second time, max diff in force= 5.330E-06 < toldff= 5.000E-05
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.72302906E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.72302906E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.33434757E-05 sigma(2 1)= 0.00000000E+00
--- !ResultsGS
iteration_state: {dtset: 3, itime: 7, icycle: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.7796667, -3.7796667, 0.0000000, ]
- [ 3.7796667, 3.7796667, 0.0000000, ]
- [ 0.0000000, 0.0000000, 26.4576669, ]
lattice_lengths: [ 5.34526, 5.34526, 26.45767, ]
lattice_angles: [ 90.000, 90.000, 90.000, ] # degrees, (23, 13, 12)
lattice_volume: 7.5594212E+02
convergence: {deltae: -1.980E-07, res2: 8.329E-07, residm: 3.687E-09, diffor: 5.330E-06, }
etotal : -1.17553431E+01
entropy : 0.00000000E+00
fermie : 1.68660635E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.72302906E-04, 0.00000000E+00, 0.00000000E+00, ]
- [ 0.00000000E+00, 1.72302906E-04, 0.00000000E+00, ]
- [ 0.00000000E+00, 0.00000000E+00, 4.33434757E-05, ]
pressure_GPa: -3.8046E+00
xred :
- [ 0.0000E+00, 0.0000E+00, -8.2502E-03, Al]
- [ -5.0000E-01, 5.0000E-01, 1.3834E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 2.8571E-01, Al]
- [ -5.0000E-01, 5.0000E-01, 4.3308E-01, Al]
- [ 0.0000E+00, 0.0000E+00, 5.7968E-01, Al]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, 3.45960549E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -2.69364718E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, 2.69364718E-04, ]
- [ -0.00000000E+00, -0.00000000E+00, -3.45960549E-04, ]
force_length_stats: {min: 0.00000000E+00, max: 3.45960549E-04, mean: 2.46130107E-04, }
...
Integrated electronic density in atomic spheres:
------------------------------------------------
Atom Sphere_radius Integrated_density
1 2.00000 0.85694733
2 2.00000 0.93453717
3 2.00000 0.92140601
4 2.00000 0.91322203
5 2.00000 0.90176526
---OUTPUT-----------------------------------------------------------------------
Cartesian coordinates (xcart) [bohr]
0.00000000000000E+00 0.00000000000000E+00 -2.18280679472094E-01
0.00000000000000E+00 3.77966669430000E+00 3.66027357974869E+00
0.00000000000000E+00 0.00000000000000E+00 7.55933338860000E+00
0.00000000000000E+00 3.77966669430000E+00 1.14583931974513E+01
0.00000000000000E+00 0.00000000000000E+00 1.53369474566721E+01
Reduced coordinates (xred)
0.00000000000000E+00 0.00000000000000E+00 -8.25018625513336E-03
-5.00000000000000E-01 5.00000000000000E-01 1.38344533518511E-01
0.00000000000000E+00 0.00000000000000E+00 2.85714285714286E-01
-5.00000000000000E-01 5.00000000000000E-01 4.33084037910061E-01
0.00000000000000E+00 0.00000000000000E+00 5.79678757683705E-01
Cartesian forces (fcart) [Ha/bohr]; max,rms= 3.45961E-04 1.60102E-04 (free atoms)
-0.00000000000000E+00 -0.00000000000000E+00 3.45960549364919E-04
-0.00000000000000E+00 -0.00000000000000E+00 -2.69364718127626E-04
-0.00000000000000E+00 -0.00000000000000E+00 -0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 2.69364718127626E-04
-0.00000000000000E+00 -0.00000000000000E+00 -3.45960549364919E-04
Gradient of E wrt nuclear positions in reduced coordinates (gred)
-0.00000000000000E+00 -0.00000000000000E+00 -9.15330896183420E-03
-0.00000000000000E+00 0.00000000000000E+00 7.12676197608547E-03
-0.00000000000000E+00 0.00000000000000E+00 0.00000000000000E+00
-0.00000000000000E+00 -0.00000000000000E+00 -7.12676197608547E-03
-0.00000000000000E+00 0.00000000000000E+00 9.15330896183420E-03
Total energy (etotal) [Ha]= -1.17553431364498E+01
Difference of energy with previous step (new-old):
Absolute (Ha)=-1.90355E-05
Relative =-1.61931E-06
At Broyd/MD step 7, gradients are converged :
max grad (force/stress) = 3.4596E-04 < tolmxf= 5.0000E-04 ha/bohr (free atoms)
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 38.157E-11; max= 36.871E-10
reduced coordinates (array xred) for 5 atoms
0.000000000000 0.000000000000 -0.008250186255
-0.500000000000 0.500000000000 0.138344533519
0.000000000000 0.000000000000 0.285714285714
-0.500000000000 0.500000000000 0.433084037910
0.000000000000 0.000000000000 0.579678757684
rms dE/dt= 4.2359E-03; max dE/dt= 9.1533E-03; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 -0.009153308962
2 0.000000000000 0.000000000000 0.007126761976
3 0.000000000000 0.000000000000 0.000000000000
4 0.000000000000 0.000000000000 -0.007126761976
5 0.000000000000 0.000000000000 0.009153308962
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 -0.11550916107869
2 0.00000000000000 2.00011347807574 1.93693336275932
3 0.00000000000000 0.00000000000000 4.00022695615147
4 0.00000000000000 2.00011347807574 6.06352054954363
5 0.00000000000000 0.00000000000000 8.11596307338164
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 0.00034596054936
2 -0.00000000000000 -0.00000000000000 -0.00026936471813
3 -0.00000000000000 -0.00000000000000 -0.00000000000000
4 -0.00000000000000 -0.00000000000000 0.00026936471813
5 -0.00000000000000 -0.00000000000000 -0.00034596054936
frms,max,avg= 1.6010249E-04 3.4596055E-04 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 0.01779000672565
2 -0.00000000000000 -0.00000000000000 -0.01385129072069
3 -0.00000000000000 -0.00000000000000 -0.00000000000000
4 -0.00000000000000 -0.00000000000000 0.01385129072069
5 -0.00000000000000 -0.00000000000000 -0.01779000672565
frms,max,avg= 8.2328010E-03 1.7790007E-02 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.559333388600 7.559333388600 7.559333388600 bohr
= 4.000226956151 4.000226956151 4.000226956151 angstroms
prteigrs : about to open file tbase4_7o_DS3_EIG
Fermi (or HOMO) energy (hartree) = 0.16866 Average Vxc (hartree)= -0.30829
Eigenvalues (hartree) for nkpt= 6 k points:
kpt# 1, nband= 11, wtk= 0.12500, kpt= -0.1250 0.0000 0.0000 (reduced coord)
-0.22225 -0.18954 -0.13522 -0.06463 0.02224 0.09681 0.19242 0.26210
0.29058 0.32482 0.33759
occupation numbers for kpt# 1
2.00000 2.00000 2.00000 2.00000 2.00001 2.06357 0.13255 0.00205
0.00012 0.00000 0.00000
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 3, itime: 7, icycle: 1, }
comment : Components of total free energy in Hartree
kinetic : 4.10659346848757E+00
hartree : 6.91334709453081E+00
xc : -5.45113046865728E+00
Ewald energy : -4.96203662478673E+00
psp_core : 5.69876881015122E-02
local_psp : -1.45440655811596E+01
non_local_psp : 2.12747145529473E+00
internal : -1.17528329681890E+01
'-kT*entropy' : -2.51016826082129E-03
total_energy : -1.17553431364498E+01
total_energy_eV : -3.19879182539977E+02
band_energy : 2.03966531477191E-01
...
rms coord change= 3.4337E-03 atom, delta coord (reduced):
1 0.000000000000 0.000000000000 -0.008250186255
2 0.000000000000 0.000000000000 -0.004512609339
3 0.000000000000 0.000000000000 -0.000000000000
4 0.000000000000 0.000000000000 0.004512609339
5 0.000000000000 0.000000000000 0.008250186255
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.72302906E-04 sigma(3 2)= 0.00000000E+00
sigma(2 2)= 1.72302906E-04 sigma(3 1)= 0.00000000E+00
sigma(3 3)= 4.33434757E-05 sigma(2 1)= 0.00000000E+00
-Cartesian components of stress tensor (GPa) [Pressure= -3.8046E+00 GPa]
- sigma(1 1)= 5.06932651E+00 sigma(3 2)= 0.00000000E+00
- sigma(2 2)= 5.06932651E+00 sigma(3 1)= 0.00000000E+00
- sigma(3 3)= 1.27520908E+00 sigma(2 1)= 0.00000000E+00
== END DATASET(S) ==============================================================
================================================================================
-outvars: echo values of variables after computation --------
acell 7.5593333886E+00 7.5593333886E+00 7.5593333886E+00 Bohr
amu 2.69815390E+01
chksymtnons 0
ecut 6.00000000E+00 Hartree
etotal1 -7.0358683757E+00
etotal2 -9.3978596458E+00
etotal3 -1.1755343136E+01
fcart1 -0.0000000000E+00 -0.0000000000E+00 -2.0972487346E-04
-0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
-0.0000000000E+00 -0.0000000000E+00 2.0972487346E-04
fcart2 -0.0000000000E+00 -0.0000000000E+00 1.6698252648E-04
-0.0000000000E+00 -0.0000000000E+00 -1.1616651849E-04
-0.0000000000E+00 -0.0000000000E+00 1.1616651849E-04
-0.0000000000E+00 -0.0000000000E+00 -1.6698252648E-04
fcart3 -0.0000000000E+00 -0.0000000000E+00 3.4596054936E-04
-0.0000000000E+00 -0.0000000000E+00 -2.6936471813E-04
-0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
-0.0000000000E+00 -0.0000000000E+00 2.6936471813E-04
-0.0000000000E+00 -0.0000000000E+00 -3.4596054936E-04
- fftalg 512
ionmov 2
iprcel 45
ixc -1012
jdtset 1 2 3
kpt -1.25000000E-01 0.00000000E+00 0.00000000E+00
-3.75000000E-01 0.00000000E+00 0.00000000E+00
-2.50000000E-01 1.25000000E-01 0.00000000E+00
5.00000000E-01 1.25000000E-01 0.00000000E+00
-3.75000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 3.75000000E-01 0.00000000E+00
kptrlatt 4 4 0 -4 4 0 0 0 1
kptrlen 3.02373336E+01
P mkmem 6
natom1 3
natom2 4
natom3 5
nband1 7
nband2 8
nband3 11
ndtset 3
ngfft1 12 12 45
ngfft2 12 12 54
ngfft3 12 12 60
nkpt 6
nstep 10
nsym 16
ntime 10
ntypat 1
occ1 2.000000 2.000000 2.000000 2.134837 0.002908 0.000192
0.000000
2.000000 2.000000 2.000696 2.016432 2.119803 0.102900
0.005244
2.000000 2.000000 2.000125 1.485023 0.830030 0.004862
0.001254
2.000000 2.000000 2.000265 2.000648 1.720901 0.594914
0.003110
2.000000 2.000001 2.038458 2.141836 1.009421 0.004559
0.002642
2.000438 2.000726 2.133857 2.121552 0.000437 0.001313
0.002407
occ2 2.000000 2.000000 2.000000 2.000001 2.044292 0.004318
0.000555 0.000271
2.000000 2.000000 2.000000 2.000169 2.028061 2.049418
1.338406 0.778421
2.000000 2.000000 2.000000 2.000911 2.087100 1.257100
0.027404 0.001665
2.000000 2.000000 2.000000 2.000000 2.011939 2.011939
0.650135 0.650135
2.000000 2.000000 2.000077 2.016934 2.136339 2.059181
0.104373 0.006144
2.000136 2.000136 2.024265 2.024265 1.486413 1.486413
0.007928 0.007928
occ3 2.000000 2.000000 2.000000 2.000000 2.000009 2.063566
0.132545 0.002053 0.000116 0.000001 0.000000
2.000000 2.000000 2.000000 2.000004 2.000017 2.001647
2.080607 2.130699 0.839788 0.253125 0.002563
2.000000 2.000000 2.000000 2.000000 2.003181 2.015735
1.818345 0.571038 0.001936 0.005039 0.000071
2.000000 2.000000 2.000000 2.000000 2.000028 2.000032
2.050528 2.091180 0.669452 0.067432 0.005183
2.000000 2.000000 2.000000 2.001710 2.003766 2.060008
1.911046 1.597650 0.010652 0.001135 0.004775
2.000012 2.000015 2.000985 2.001739 2.107353 2.115040
0.710996 0.440069 0.125822 0.094627 0.000591
occopt 4
optforces 1
rprim1 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 2.5000000000E+00
rprim2 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 3.0000000000E+00
rprim3 5.0000000000E-01 -5.0000000000E-01 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 3.5000000000E+00
shiftk 5.00000000E-01 5.00000000E-01 0.00000000E+00
spgroup1 123
spgroup2 129
spgroup3 123
strten1 1.9309479760E-04 1.9309479760E-04 1.9597988539E-05
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten2 2.1258114619E-04 2.1258114619E-04 8.5558596550E-05
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten3 1.7230290577E-04 1.7230290577E-04 4.3343475651E-05
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
-1 0 0 0 1 0 0 0 -1 1 0 0 0 -1 0 0 0 1
-1 0 0 0 -1 0 0 0 1 1 0 0 0 1 0 0 0 -1
1 0 0 0 -1 0 0 0 -1 -1 0 0 0 1 0 0 0 1
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 -1 0 1 0 0 0 0 -1 0 1 0 -1 0 0 0 0 1
0 -1 0 -1 0 0 0 0 1 0 1 0 1 0 0 0 0 -1
0 1 0 -1 0 0 0 0 -1 0 -1 0 1 0 0 0 0 1
tnons1 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4000000
0.0000000 0.0000000 0.4000000 0.0000000 0.0000000 0.0000000
tnons2 0.0000000 0.0000000 0.0000000 0.5000000 0.5000000 0.5000000
0.5000000 0.5000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.5000000 0.5000000 0.5000000
0.5000000 0.5000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.5000000 0.5000000 0.5000000
0.5000000 0.5000000 0.5000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.5000000 0.5000000 0.5000000
0.5000000 0.5000000 0.5000000 0.0000000 0.0000000 0.0000000
tnons3 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 -0.4285714
0.0000000 0.0000000 -0.4285714 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 -0.4285714
0.0000000 0.0000000 -0.4285714 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 -0.4285714
0.0000000 0.0000000 -0.4285714 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 -0.4285714
0.0000000 0.0000000 -0.4285714 0.0000000 0.0000000 0.0000000
toldff 5.00000000E-05
tolmxf 5.00000000E-04
tsmear 4.00000000E-02 Hartree
typat1 1 1 1
typat2 1 1 1 1
typat3 1 1 1 1 1
wtk 0.12500 0.12500 0.25000 0.12500 0.25000 0.12500
xangst1 0.0000000000E+00 0.0000000000E+00 -9.5791295042E-02
0.0000000000E+00 2.0001134781E+00 2.0001134781E+00
0.0000000000E+00 0.0000000000E+00 4.0960182512E+00
xangst2 0.0000000000E+00 0.0000000000E+00 -1.8763407917E-01
0.0000000000E+00 2.0001134781E+00 1.9285500770E+00
0.0000000000E+00 0.0000000000E+00 4.0717903572E+00
0.0000000000E+00 2.0001134781E+00 6.1879745134E+00
xangst3 0.0000000000E+00 0.0000000000E+00 -1.1550916108E-01
0.0000000000E+00 2.0001134781E+00 1.9369333628E+00
0.0000000000E+00 0.0000000000E+00 4.0002269562E+00
0.0000000000E+00 2.0001134781E+00 6.0635205495E+00
0.0000000000E+00 0.0000000000E+00 8.1159630734E+00
xcart1 0.0000000000E+00 0.0000000000E+00 -1.8101931288E-01
0.0000000000E+00 3.7796666943E+00 3.7796666943E+00
0.0000000000E+00 0.0000000000E+00 7.7403527015E+00
xcart2 0.0000000000E+00 0.0000000000E+00 -3.5457702151E-01
0.0000000000E+00 3.7796666943E+00 3.6444314657E+00
0.0000000000E+00 0.0000000000E+00 7.6945686172E+00
0.0000000000E+00 3.7796666943E+00 1.1693577104E+01
xcart3 0.0000000000E+00 0.0000000000E+00 -2.1828067947E-01
0.0000000000E+00 3.7796666943E+00 3.6602735797E+00
0.0000000000E+00 0.0000000000E+00 7.5593333886E+00
0.0000000000E+00 3.7796666943E+00 1.1458393197E+01
0.0000000000E+00 0.0000000000E+00 1.5336947457E+01
xred1 0.0000000000E+00 0.0000000000E+00 -9.5785860245E-03
-5.0000000000E-01 5.0000000000E-01 2.0000000000E-01
0.0000000000E+00 0.0000000000E+00 4.0957858602E-01
xred2 0.0000000000E+00 0.0000000000E+00 -1.5635286133E-02
-5.0000000000E-01 5.0000000000E-01 1.6070338827E-01
0.0000000000E+00 0.0000000000E+00 3.3929661173E-01
-5.0000000000E-01 5.0000000000E-01 5.1563528613E-01
xred3 0.0000000000E+00 0.0000000000E+00 -8.2501862551E-03
-5.0000000000E-01 5.0000000000E-01 1.3834453352E-01
0.0000000000E+00 0.0000000000E+00 2.8571428571E-01
-5.0000000000E-01 5.0000000000E-01 4.3308403791E-01
0.0000000000E+00 0.0000000000E+00 5.7967875768E-01
znucl 13.00000
================================================================================
- Timing analysis has been suppressed with timopt=0
================================================================================
Suggested references for the acknowledgment of ABINIT usage.
The users of ABINIT have little formal obligations with respect to the ABINIT group
(those specified in the GNU General Public License, http://www.gnu.org/copyleft/gpl.txt).
However, it is common practice in the scientific literature,
to acknowledge the efforts of people that have made the research possible.
In this spirit, please find below suggested citations of work written by ABINIT developers,
corresponding to implementations inside of ABINIT that you have used in the present run.
Note also that it will be of great value to readers of publications presenting these results,
to read papers enabling them to understand the theoretical formalism and details
of the ABINIT implementation.
For information on why they are suggested, see also https://docs.abinit.org/theory/acknowledgments.
-
- [1] Libxc: A library of exchange and correlation functionals for density functional theory.
- M.A.L. Marques, M.J.T. Oliveira, T. Burnus, Computer Physics Communications 183, 2227 (2012).
- Comment: to be cited when LibXC is used (negative value of ixc)
- Strong suggestion to cite this paper.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#marques2012
-
- [2] Preconditioning of self-consistent-field cycles in density functional theory: the extrapolar method
- P.-M. Anglade, X. Gonze, Phys. Rev. B 78, 045126 (2008).
- Comment: to be cited in case the extrapolar conditioner is used, i.e. non-vanishing iprcel.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#anglade2008
-
- [3] The Abinit project: Impact, environment and recent developments.
- Computer Phys. Comm. 248, 107042 (2020).
- X.Gonze, B. Amadon, G. Antonius, F.Arnardi, L.Baguet, J.-M.Beuken,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, N.Brouwer, F.Bruneval,
- G.Brunin, T.Cavignac, J.-B. Charraud, Wei Chen, M.Cote, S.Cottenier,
- J.Denier, G.Geneste, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, Xu He, N.Helbig, N.Holzwarth, Y.Jia, F.Jollet,
- W.Lafargue-Dit-Hauret, K.Lejaeghere, M.A.L.Marques, A.Martin, C.Martins,
- H.P.C. Miranda, F.Naccarato, K. Persson, G.Petretto, V.Planes, Y.Pouillon,
- S.Prokhorenko, F.Ricci, G.-M.Rignanese, A.H.Romero, M.M.Schmitt, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, G.Zerah and J.W.Zwanzig
- Comment: the fifth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT20.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2020
-
- [4] Optimized norm-conserving Vanderbilt pseudopotentials.
- D.R. Hamann, Phys. Rev. B 88, 085117 (2013).
- Comment: Some pseudopotential generated using the ONCVPSP code were used.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#hamann2013
-
- [5] ABINIT: Overview, and focus on selected capabilities
- J. Chem. Phys. 152, 124102 (2020).
- A. Romero, D.C. Allan, B. Amadon, G. Antonius, T. Applencourt, L.Baguet,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, F.Bruneval,
- G.Brunin, D.Caliste, M.Cote,
- J.Denier, C. Dreyer, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, F.Jollet, G. Jomard,
- A.Martin,
- H.P.C. Miranda, F.Naccarato, G.Petretto, N.A. Pike, V.Planes,
- S.Prokhorenko, T. Rangel, F.Ricci, G.-M.Rignanese, M.Royo, M.Stengel, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, J.Wiktor, J.W.Zwanziger, and X.Gonze.
- Comment: a global overview of ABINIT, with focus on selected capabilities .
- Note that a version of this paper, that is not formatted for J. Chem. Phys
- is available at https://www.abinit.org/sites/default/files/ABINIT20_JPC.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#romero2020
-
- [6] Recent developments in the ABINIT software package.
- Computer Phys. Comm. 205, 106 (2016).
- X.Gonze, F.Jollet, F.Abreu Araujo, D.Adams, B.Amadon, T.Applencourt,
- C.Audouze, J.-M.Beuken, J.Bieder, A.Bokhanchuk, E.Bousquet, F.Bruneval
- D.Caliste, M.Cote, F.Dahm, F.Da Pieve, M.Delaveau, M.Di Gennaro,
- B.Dorado, C.Espejo, G.Geneste, L.Genovese, A.Gerossier, M.Giantomassi,
- Y.Gillet, D.R.Hamann, L.He, G.Jomard, J.Laflamme Janssen, S.Le Roux,
- A.Levitt, A.Lherbier, F.Liu, I.Lukacevic, A.Martin, C.Martins,
- M.J.T.Oliveira, S.Ponce, Y.Pouillon, T.Rangel, G.-M.Rignanese,
- A.H.Romero, B.Rousseau, O.Rubel, A.A.Shukri, M.Stankovski, M.Torrent,
- M.J.Van Setten, B.Van Troeye, M.J.Verstraete, D.Waroquier, J.Wiktor,
- B.Xu, A.Zhou, J.W.Zwanziger.
- Comment: the fourth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT16.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2016
-
- Proc. 0 individual time (sec): cpu= 22.6 wall= 22.8
================================================================================
Calculation completed.
.Delivered 33 WARNINGs and 4 COMMENTs to log file.
+Overall time at end (sec) : cpu= 22.6 wall= 22.8
This run might take about one minute, and is the longest of the four basic tutorials. You should start it now.
You will notice that the SCF convergence is rather satisfactory, for all the cases (3, 4 or 5 metal layers).
For the 3 aluminum layer case, one has the non-relaxed total energy:
ETOT 6 -7.0350153035193
(this quantity is converged, unlike in test 4.6) giving the unrelaxed surface energy 0.0193 Ha = 0.525 eV; and for the relaxed case:
etotal1 -7.0358683757E+00
(by contrast the difference with test 4.6 is less than 1 microHa) giving the relaxed surface energy 0.0189 Ha = 0.514 eV.
For the 4 aluminum layer case, one has the non-relaxed total energy:
ETOT 6 -9.3958299123967
giving the unrelaxed surface energy 0.0178 Ha = 0.484 eV; and for the relaxed case:
etotal2 -9.3978596458E+00
giving the relaxed surface energy 0.0168 Ha = 0.457 eV.
For the 5 aluminum layer case, one has the non-relaxed total energy:
ETOT 6 -11.754755842794
giving the unrelaxed surface energy 0.0173 Ha = 0.471 eV; and for the relaxed case:
etotal3 -1.1755343136E+01
giving the relaxed surface energy 0.0170 Ha = 0.463 eV.
The relative difference in the surface energy of the 4 and 5 layer cases is on the order of 1.2%.
In the framework of this tutorial, we will not pursue this investigation, which is a simple application of the concepts already explored.
Just for your information, and as an additional warning, when the work accomplished until now is completed with 6 and 7 layers without relaxation (see $ABI_TESTS/tutorial/Input/tbase4_8.abi and $ABI_TESTS/tutorial/Refs/tbase4_8.abo where 5, 6 and 7 layers are treated), this non-relaxed energy surface energy behaves as follows:
| number of aluminum layers | surface energy |
|---|---|
| 3 | 0.525 eV |
| 4 | 0.484 eV |
| 5 | 0.471 eV |
| 6 | 0.419 eV |
| 7 | 0.426 eV |
So, the surface energy convergence is rather difficult to reach. Our values, with a 4x4x1 grid,
a smearing of 0.04 Ha, a kinetic energy cut-off of 6 Ha, the Al.psp8 pseudopotential,
still oscillate between 0.42 eV and 0.53 eV.
Increasing the k-point sampling might decrease slightly the oscillations, but note that this effect
is intrinsic to the computation of properties of a metallic surface: the electrons are confined inside the slab potential,
with sub-bands in the direction normal to the surface, and the Fermi energy oscillates with the width of the slab.
This effect might be understood based on a comparison with the behaviour of a jellium slab.
An error on the order of 0.019 eV is due to the thin vacuum layer.
Other sources of errors might have to be rechecked, seeing the kind of accuracy that is needed.
Experimental data give a surface energy around 0.55 eV (sorry, the reference is to be provided).
Soft and hard pseudopotentials¶
In the context of a plane-wave basis, a soft pseudopotential means that a low ecut will be required to obtain convergence whereas a hard pseudopotential implies that a high ecut will be needed. It can be understood by considering the pseudo-wave-functions of that atom. A hard pseudopotential has pseudo-wave-functions that have sharp features in real space which require many plane-waves to describe.
On the other hand, a soft pseudopotential has rather smooth pseudo-wave-functions. They need fewer plane-waves to be described precisely than the pseudo-wave-functions of hard pseudopotentials. This designation is somewhat qualitative, and it is relative to other pseudopotentials. In other words, a pseudopotential can be soft when compared to a certain pseudopotential but hard with respect to another.
In general, pseudopotentials describing light elements, those of the 2nd line of the periodic table, and pseudopotentials that include semi-core states are considered hard as they have strongly peaked pseudo-wave-functions that require a large ecut. This discussion is valid for norm-conserving pseudopotentials. With PAW pseudopotentials, we are able to keep pseudo-wave-function smooth which means that they will require lower ecut than their norm-conserving counterpart which is one of their main benefits.