Hubbard U and Hund’s J Parameters with Cococcioni and de Gironcoli’s approach¶
1 How to determine U and/or J for DFT+U(+J) via Linear Response¶
This tutorial aims to demonstrate the operations and functionalities of the Abinit post-processing utility called Linear Response Hubbard U and Hund’s J (lruj), designed to determine the first-principles Hubbard U and/or Hund’s J parameters for particular atomic subspaces. Once obtained, these parameters may then be applied via the DFT+U(+J)-like Hubbard functionals to address self-interaction and static correlation errors.
Note that there is another methodology to compute U and J; see the cRPA U(J) tutorial.
This tutorial is a condensed version of that published in [MacEnulty2024], a more comprehensive user guide on the lrUJ utility and its predessecor, the UJdet utility. If you find this information useful for your own scientific investigations, please cite [MacEnulty2024].
In this tutorial, you will learn how to run perturbative calculations in Abinit and generate input data to successfully execute the lruj post-processing utility. We strongly encourage you to read the PAW1, PAW2 and DFT+U tutorials to familiarize yourself with the manifestation of PAW atomic datasets within Abinit. Also consider checking out the following video introducing the PAW formalism in an Abinit context.
This tutorial should take less than 30 minutes. We begin with a brief description of the linear response method and an important explanation of recent renovations to the linear response functionalities of Abinit. Click here if you’d like to skip to the NiO tutorial directly.
Note
Supposing you made your own installation of ABINIT, the input files to run the examples are in the ~abinit/tests/ directory where ~abinit is the absolute path of the abinit top-level directory. If you have NOT made your own install, ask your system administrator where to find the package, especially the executable and test files.
In case you work on your own PC or workstation, to make things easier, we suggest you define some handy environment variables by executing the following lines in the terminal:
export ABI_HOME=Replace_with_absolute_path_to_abinit_top_level_dir # Change this line
export PATH=$ABI_HOME/src/98_main/:$PATH # Do not change this line: path to executable
export ABI_TESTS=$ABI_HOME/tests/ # Do not change this line: path to tests dir
export ABI_PSPDIR=$ABI_TESTS/Pspdir/ # Do not change this line: path to pseudos dir
Examples in this tutorial use these shell variables: copy and paste
the code snippets into the terminal (remember to set ABI_HOME first!) or, alternatively,
source the set_abienv.sh script located in the ~abinit directory:
source ~abinit/set_abienv.sh
The ‘export PATH’ line adds the directory containing the executables to your PATH so that you can invoke the code by simply typing abinit in the terminal instead of providing the absolute path.
To execute the tutorials, create a working directory (Work*) and
copy there the input files of the lesson.
Most of the tutorials do not rely on parallelism (except specific tutorials on parallelism). However you can run most of the tutorial examples in parallel with MPI, see the topic on parallelism.
2 Summary of linear response method to determine U(J)¶
The Hubbard U and Hund’s J are ground-state properties of any multi-atomic system treated with a given approximate XC functional. They embody the spurious curvature of the total energy with respect to subspace occupation and magnetization respectively. The Hubbard U, specifically, is compensating for an unphysical quadratic term left over by the Hartree energy; it is thus defined as the second derivative of the total energy with respect to charge occupation: \(U=\frac{\delta^2 E}{\delta^2 n}\).
Cococcioni and de Gironcoli [Cococcioni2002], following the seminal work from Pickett et al. in 1998 [Pickett1998], further defined a protocol to strictly avoid the semi-empirical evaluation of these parameters. This linear response procedure is described mathematically in terms of constraint formalism and Lagrange coefficients by Dederichs et al. [Dederichs1984] and Anisimov et al. [Anisimov1991]. A formal definition of and analogous procedure for the Hund’s coupling J parameter was published by Linscott et al. in 2018 [Linscott2018].
Practically, the linear response approach begins with the application of a small perturbation \(\alpha\)(\(\beta\)) to the external potential of the subspace for which U(J) is under assessment. The change in electronic occupation induced by that perturbation is then monitored. This occupational response is, for small perturbations, expected to be a linear function of the perturbation’s magnitude.
Important
For more detailed information on the concepts of the linear response calculation of the Hubbard U and Hund’s J parameters, please see the following papers.
[1] “Linear response approach to the calculation of the effective interaction parameters in the LDA + U method”, M. Cococcioni and S. de Gironcoli, Physical Review B 71, 035105 (2005) [Cococcioni2005]
[2] “The role of spin in the calculation of Hubbard U and Hund’s J parameters from first principles”, E.B. Linscott, D.J. Cole, M.C. Payne and D.D. O’Regan, Physical Review B 98, 235157 (2018) [Linscott2018]
[3] “A LDA+U study of selected iron compounds “, M. Cococcioni, Ph.D. thesis, International School for Advanced Studies (SISSA), Trieste (2002) [Cococcioni2002]
Some further reading:
[4] “Ground States of Constrained Systems: Application to Cerium Impurities”, P. H. Dederichs, S. Blugel, R. Zeller, and H. Akai, Phys. Rev. Lett. 53, 2512 (1984) [Dederichs1984]
[5] “Calculation of Coulomb-interaction parameters for La2CuO4 using a constrained-density-functional approach”, M. S. Hybertsen, M. Schluter, and N. E. Christensen, Phys. Rev. B 39, 9028 (1989) [Hybertsen1989]
[6] “Density-functional calculation of effective Coulomb interactions in metals”, V. I. Anisimov and O. Gunnarsson, Phys. Rev. B42, 7570 (1991) [Anisimov1991]
[7] “Reformulation of the LDA+U method for a local-orbital basis”, W. E. Pickett, S. C. Erwin, and E. C. Ethridge, Phys. Rev. B58, 1201 (1998) [Pickett1998]
The implementation of the determination of U in ABINIT is briefly discussed in [Gonze2016].
3 Renovation of the linear response protocol since Abinit Version 9.6.2¶
The basic functions of linear response were implemented in Abinit, embedded in its PAW functionality, in 2009. The subsequent U(J) DETermination (ujdet) program was part of the Abinit suite as both an intrinsic DFT protocol and post-processing utility extension. Abinit Version 9.6.2 saw the introduction of an additional macro_uj variable for calculating the Hund’s J with ujdet.
In 2022, users alerted Abinit to the existence of a bug in ujdet which led to the decommission of its post-processing utility and the renovation of its internal Abinit functionality. As of Version 9.6.2, the Abinit DFT suite is equipped with the lruj post-processing tool, which is built off of the same core, but debugged, ujdet programming. Although older versions of Abinit preserve the ujdet internal and post-processing utilities, their use is strongly disadvised. The lruj functionality conserves most of ujdet’s data processing functionalities. For retrogressive and archive purposes, the primary differences between the two are outlined in the table below.
ujdet |
lruj |
|
|---|---|---|
| 1 | Embedded in Abinit core routine + Post-processing extension | Post-processor |
| 2 | Two-point linear regression | 3+ point polynomial (variable degree) regression |
| 3 | \(\chi\) and \(\chi_0\) responses treated as matrices; interatomic response monitored; matrices augmented by total system charge | \(\chi\) and \(\chi_0\) responses treated as scalars |
| 4 | Supercell extrapolation scheme | RMS Error analysis |
| 5 | Atomic Sphere Approximation projector extensions/normalizations |
As mentioned in item (2), the most influential difference between ujdet and lruj is the number of data points used to compute a linear regression of the response functions \(\chi\) and \(\chi_0\). The ujdet utility uses only two points: the unperturbed case—in which the perturbation applied is zero and the subspace occupations are those of the ground state—and one perturbed case, in which the potential perturbation is equal to the value of pawujv. Note that the ujdet procedure differs slightly from its implementations in Abinit versions prior to 9.6.2, in which it conducted two perturbations: one of strength pawujv and the other of strength \(-1.0*\)pawujv. Due to a bug in the program, the second perturbation administered provided erroneous unscreened response occupations. To fix this, we exchanged the data point from the second perturbation for one from the unperturbed case, whose occupations are calculated anyway from the ground state wavefunctions read into Abinit.
By contrast, the lruj utility requires, at minimum, three data points (one unperturbed case and at least two perturbations) to conduct a distinctive regression analysis on the response functions. With n data points, the lruj utility computes not only a linear regression, but all polynomial regressions up to degree \(n-2\). Furthermore, the lruj utility conducts RMS error analysis on the fits and factors that into an approximative RMS error on the resulting Hubbard parameters.
Another crucial difference between the two utilities is item (3) in the above table: the ujdet utility treats the response functions as matrices, whereas the lruj utility treats them as scalars. Ideologically, this means that the ujdet Hubbard parameters are, to some degree, informed by the Hubbard interactions on and between the other atomic subspaces of the system as well as the total charge bath. The protocol is expanded upon in reference [Cococcioni2002].
By contrast, the lruj utility provides the scalar Hubbard parameters, informed only by the change in occupancy on the perturbed subspace. This parameter is functionally sufficient for SIE corrective application to that subspace.
For all other purposes, it can be said that lruj offers a simplified data processing procedure to that of ujdet. More detailed information is due to follow in the coming months in the form of a user guide, so stay tuned.
4 Determine the Hubbard U for Ni 3d in NiO with lruj ¶
For this tutorial, we will calculate the scalar Hubbard U parameter for the Ni 3d subspace in a four-atom unit cell of AF2-ordered NiO using the lruj post-processing utility. The lruj procedure can be carried out in three steps:
- Run a ground state Abinit calculation for NiO to generate
WFKfiles. - Run a series of perturbative Abinit calculations to generate
*_LRUJ.ncfiles. - Execute the lruj prost-processing utility.
4.1. Generate the ground-state WFK files¶
We need to establish a ground state material system whose subspace potential we can perturb.
Fortunately, this is principally no different from your standard Abinit ground state run, aside
from a few minor modifications to the input file. Make a new work directory called
work_lruj, then copy and paste tlruj_1.abi and tlruj_2.abi
therein:
cd $ABI_TESTS/tutorial/Input
mkdir work_lruj
cd work_lruj
cp ../tlruj_1.abi .
cp ../tlruj_2.abi .
Important
Henceforth, the name of files are mentioned as if you were in such a subdirectory. All the input files can be found in the $ABI_TESTS/tutorial/Input directory. You can compare your results with the reference output files located in $ABI_TESTS/tutorial/Refs directory (for the prese
Open up tlruj_1.abi; you’ll notice a few key differences between this and other
standard ground-state input files.
################################################################# ## Automatic test/tutorial for ABINIT: ## ## Linear Response Hubbard U and Hund's J (LRUJ) ## ## post-processing utility for the determination of ## ## said parameters in situ and ab initio ## ## Initialization of WFK files ## ## ## ## Antiferro. II NiO, 4 atom | Hubbard U on Ni 3d | LMac 2022 ## ################################################################# #Run Parameters nstep 30 #Higher than normal because of magnetic state tolvrs 10d-12 ecut 10 pawecutdg 20 chkprim 0 #Will complain otherwise with AFII magnetic state occopt 0 nband 26 #24 occupied + 2 unoccupied occ 24*1.0 2*0.0 24*1.0 2*0.0 #Structural Parameters natom 4 #NOTE: Converge U(J) wrt supercell size! ntypat 3 #Specify perturbed Ni atom as a separate species typat 1 2 3 3 #to that of the other Ni atom. znucl 28 28 8 #First two are Ni atoms, last two are O acell 3*7.8800 xred 0.0000000000 0.0000000000 0.0000000000 0.5000000000 0.5000000000 0.5000000000 0.2500000000 0.2500000000 0.2500000000 0.7500000000 0.7500000000 0.7500000000 rprim 0.5000000000 0.5000000000 1.0000000000 1.0000000000 0.5000000000 0.5000000000 0.5000000000 1.0000000000 0.5000000000 #Spin Parameters nsppol 2 nspden 2 nspinor 1 spinat 0 0 3 #Set high to enforce convergence to high magnetic state 0 0 -3 #Otherwise, it may collapse to non-magnetic state 0 0 0 0 0 0 #Kpoint Grid kptopt 1 #Monkhorst-Pack Mesh chksymbreak 0 #Don't check for symmetry breaking ngkpt 4 4 4 # DFT+U usepawu 1 #Alert Abinit to use of DFT+U lpawu 2 2 1 #Subspaces of interest: Ni 3d, O 2p upawu 0.0 0.0 0.0 eV #Raw (non-corrected) XC functional required to establish U(J) jpawu 0.0 0.0 0.0 eV dmatpuopt 3 #Printing Info outdata_prefix = "tlruj_1.o" prtwf 1 #Default: make sure WFK are printed #Pseudos pp_dirpath "$ABI_PSPDIR/Psdj_paw_pbe_std/" pseudos "Ni.xml,Ni.xml,O.xml" #Use same pseudopotential for both Ni atoms ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = tlruj_1.abi, tlruj_2.abi, tlruj_3.abi #%% [files] #%% files_to_test = #%% tlruj_1.abo, tolnlines = 20, tolabs = 5.000e-05, tolrel = 0.07, fld_options = -medium #%% output_file = "tlruj_1.abo" #%% [paral_info] #%% max_nprocs = 20 #%% [extra_info] #%% authors = L. MacEnulty #%% keywords = PAW,DFTU,LRUJ,Hubbard-U,Hunds-J #%% description = #%% AFII NiO 4-atom cell | LRUJ utility | LMac 2022 #%% Linear Response Hubbard U and Hund's J (LRUJ) #%% post-processing utility for the determination of #%% said parameters in situ and ab initio #%%<END TEST_INFO>
First, we specify as a separate species the atom whose subspace we wish to apply a potential perturbation. This will alert Abinit that we want to allow the perturbed Ni 3d subspace to vary its properties independently to the other Ni atom in the cell.
Note
By specifying the perturbed atom as a separate species, Abinit will only harvest the changes in occupation of the perturbed atom. This information is sufficient for the lruj procedure, but not for ujdet. To avail of the supercell extrapolation technique, you will need to set the symmetry relations symrel explicitly. This will tell Abinit that (1) the perturbed atom should vary independently to its kin and (2) it should still collect occupation information for all atoms containing the subspace to be treated, not just that of the perturbed atom. The ujdet utility then uses these interatomic response matrix elements to inform its Hubbard parameters.
You can generate these symmetries in a separate run, wherein you specify the atom upon which the perturbation is to be applied as a different species. From the output, you read the number of symmetries (nsym), the symmetry operations (symrel) and the non-symmorphic vectors (tnons).
Here, we have a four-atom unit cell of NiO. Unlike the ujdet utility, the lruj utility can accomodate the perturbation of any Ni atom in the cell. For right now, we apply a perturbation to the first Ni atom by making the following modifications:
Original Modified
------------------------------- -------------------------------
ntypat = 2 ntypat = 3
typat = 1 1 2 2 typat = 1 2 3 3
znucl = 28 8 znucl = 28 28 8
pseudos = “Ni.xml,O.xml” pseudos = “Ni.xml,Ni.xml,O.xml”
If you are using other input variables whose dimensions are set by ntypat, you will need to change those as well.
Note
If using the ujdet functionality that performs the supercell extrapolation schema, the perturbation should only be applied to the first atom listed in xred. Make sure that the coordinates of the perturbed atom are listed first.
Since the Hubbard parameters are ground state properties of your choice XC functional, it is discouraged to make this a DFT+U(J) run in which U(J) are non-zero. Therefore, we add the following to the script
usepawu 1
lpawu 2 2 1
upawu 0.0 0.0 0.0 eV
jpawu 0.0 0.0 0.0 eV
and ensure that prtwf is set to 1 so that the WFK file is printed. The lpawu
2 setting specifies the \(3d\) orbitals on Ni as those for which we will calculate and apply U.
Other related variables include a high tolvrs = 10d-11 so that we can converge the electronic
structure to a high degree of accuracy. The ecut is chosen to be very low in order to
accelerate calculations, so raise this for precision calculations. All other variables used to
conduct a ground state calculation remain unmodified. Launch the Abinit run to print out the
WFK file.
abinit tlruj_1.abi > tlruj_1.log
This should take about a minute to run, but times vary depending on your hardware. This concludes Step 1.
4.2. Linear response perturbations and generation of LRUJ.nc files¶
Once we have our reference wavefunctions, we can start the linear response procedure, which we
summon and guide via the following additional input variables in tlruj_2.abi:
- pawujv => Strength of the perturbation (usually on the order of 10e-1 to 10e-2). Default value is 0.1 eV. (Our tests show that 0.1 eV is the optimal value, but the linear response is linear in a wide range (1-0.001 eV).)
- macro_uj => With nsppol, which parameter is Abinit to determine? For nsppol = 2, set macro_uj to 1 for Hubbard U, or set macro_uj to 4 for the Hund’s J.
It is typically enough to make macro_uj non-zero. To run a perturbative
calculation for the Hubbard U parameter, we set macro_uj to 1 and nsppol to 2. Note
also, that the irdwfk 1 and the tolvrs 1d-8 do not need to be set explicitly because
they are the defaults with a non-zero macro_uj. Lastly, ensure that the variable
pawujat, which identifies the perturbed atom, is set to the same atom specified as a
separate species in generating the WFK file.
Note
When irdwfk is set to 1, Abinit is instructed to read in the WFK files for a prior run given the files are named according to a specific convention. Alternatively, we can specify the WFK file and its path by name with the following:
irdwfk 0
getwfk_filepath "</pathtofile/filename_WFK>"
In changing only these variables, we set up only one perturbative calculation. This is sufficient to avail of the ujdet utility functionalities, which require only two data points as discussed above. However, in many, if not all, cases, one perturbation is inadequate to compute a good regression of the linear response data, and no error analysis can be conducted thereof.
For this reason, we will need to conduct several (at minimum four, although the more, the better)
perturbative calculations. We will take advantage of Abinit’s dataset function to get our system
to iteratively undergo n perturbations by setting ndtset to n and then specifying which
perturbation strengths pawujv1, pawujv2, … , pawujvn
we would like to apply. In this tutorial specifically, we take n to be 5 and vary the perturbation
magnitude between -0.15 eV and 0.10 eV. Take a look at tlruj_2.abi to visualize this
description.
################################################################# ## Automatic tutorial for ABINIT: ## ## Linear Response Hubbard U and Hund's J (LRUJ) ## ## post-processing utility for the determination of ## ## said parameters in situ and ab initio ## ## Initialization of WFK files ## ## ## ## Antiferro. II NiO, 4 atom | Hubbard U on Ni 3d | LMac 2022 ## ################################################################# #Run Parameters nstep 30 #Higher than normal because of magnetic state tolvrs 10d-10 ecut 10 pawecutdg 20 chkprim 0 #Will complain otherwise with AFII magnetic state occopt 0 nband 26 #24 occupied + 2 unoccupied occ 24*1.0 2*0.0 24*1.0 2*0.0 #Structural Parameters natom 4 #NOTE: Converge U(J) wrt supercell size! ntypat 3 #Specify perturbed Ni atom as a separate species typat 1 2 3 3 #to that of the other Ni atom. znucl 28 28 8 #First two are Ni atoms, last two are O acell 3*7.8800 xred 0.0000000000 0.0000000000 0.0000000000 0.5000000000 0.5000000000 0.5000000000 0.2500000000 0.2500000000 0.2500000000 0.7500000000 0.7500000000 0.7500000000 rprim 0.5000000000 0.5000000000 1.0000000000 1.0000000000 0.5000000000 0.5000000000 0.5000000000 1.0000000000 0.5000000000 #Spin Parameters nsppol 2 nspden 2 nspinor 1 spinat 0 0 3 #Set high to enforce convergence to high magnetic state. 0 0 -3 #Otherwise, it may collapse to non-magnetic state. 0 0 0 0 0 0 #Kpoint Grid kptopt 1 #Monkhorst-Pack Mesh chksymbreak 0 #Don't check for symmetry breaking. ngkpt 4 4 4 #DFT+U usepawu 1 #Alert Abinit to use of DFT+U. lpawu 2 2 1 #Subspaces of interest: Ni 3d, O 2p upawu 0.0 0.0 0.0 eV #Raw (non-corrected) XC functional required to establish U(J). jpawu 0.0 0.0 0.0 eV #Linear Response ndtset 5 #Six perturbations => Seven LR data points pawujat 1 #Label of perturbed atom pawujv1 -0.15 eV #List of perturbation strengths pawujv2 -0.10 eV pawujv3 -0.05 eV pawujv4 0.05 eV pawujv5 0.10 eV macro_uj 1 #Turn on linear response functionality for Hund's J dmatpuopt 3 #PAW projector for density matrix #Printing Info outdata_prefix = "tlruj_2.o" prtwf 0 #Don't print WFK prteig 0 #Don't print eigenvalues prtden 0 #Don't print density prtgsr 0 #Don't print GSR prtebands 0 #Don't print ebands #Input File Information irdwfk 0 getwfk_filepath "./tlruj_1.o_WFK" #Specify name and location of WFK file pp_dirpath "$ABI_PSPDIR/Psdj_paw_pbe_std" pseudos "Ni.xml,Ni.xml,O.xml" #Use same pseudopotential for both Ni atoms. ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = tlruj_1.abi,tlruj_2.abi,tlruj_3.abi #%% [files] #%% files_to_test = #%% tlruj_2.abo, tolnlines = 118, tolabs = 1.000e-04, tolrel = 0.025, fld_options = -medium #%% output_file = "tlruj_2.abo" #%% [paral_info] #%% max_nprocs = 20 #%% [extra_info] #%% authors = L. MacEnulty #%% keywords = PAW,DFTU,LRUJ,Hunds-J #%% description = #%% AFII NiO 4-atom cell | LRUJ utility | LMac 2022 #%% Linear Response Hubbard U and Hund's J (LRUJ) #%% post-processing utility for the determination of #%% said parameters in situ and ab initio #%%<END TEST_INFO>
To manage the print volume related to the ujdet functions, set pawprtvol and prtvol. All
other variables remain unmodified. Launch the Abinit run to print out the *_LRUJ.nc
files.
abinit tlruj_2.abi > tlruj_2.log
This more expensive calculation should take under five minutes to run. Open up the output file.
.Version 10.6.4.5 of ABINIT, released Dec 2025.
.(MPI version, prepared for a x86_64_linux_gnu13.2 computer)
.Copyright (C) 1998-2026 ABINIT group .
ABINIT comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Wed 4 Mar 2026.
- ( at 12h41 )
- input file -> /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/TestBot_MPI1/tutorial_tlruj_1-tlruj_2-tlruj_3/tlruj_2.abi
- output file -> /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/TestBot_MPI1/tutorial_tlruj_1-tlruj_2-tlruj_3/tlruj_2.abo
- root for input files -> tlruj_2i
- root for output files -> tlruj_2.o
- inpspheads : Reading pseudopotential header in XML form from
- /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/Ni.xml
- inpspheads : Reading pseudopotential header in XML form from
- /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/Ni.xml
- inpspheads : Reading pseudopotential header in XML form from
- /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/O.xml
DATASET 1 : space group R-3 m (#166); Bravais hR (rhombohedral)
================================================================================
Values of the parameters that define the memory need for DATASET 1.
intxc = 0 ionmov = 0 iscf = 17 lmnmax = 18
lnmax = 6 mgfft = 30 mpssoang = 3 mqgrid = 3001
natom = 4 nloc_mem = 2 nspden = 2 nspinor = 1
nsppol = 2 nsym = 12 n1xccc = 1 ntypat = 3
occopt = 0 xclevel = 2
- mband = 26 mffmem = 1 mkmem = 10
mpw = 376 nfft = 27000 nkpt = 10
PAW method is used; the additional fine FFT grid is defined by:
mgfftf= 40 nfftf = 64000
================================================================================
P This job should need less than 41.508 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 2.985 Mbytes ; DEN or POT disk file : 0.979 Mbytes.
================================================================================
DATASET 2 : space group R-3 m (#166); Bravais hR (rhombohedral)
================================================================================
Values of the parameters that define the memory need for DATASET 2.
intxc = 0 ionmov = 0 iscf = 17 lmnmax = 18
lnmax = 6 mgfft = 30 mpssoang = 3 mqgrid = 3001
natom = 4 nloc_mem = 2 nspden = 2 nspinor = 1
nsppol = 2 nsym = 12 n1xccc = 1 ntypat = 3
occopt = 0 xclevel = 2
- mband = 26 mffmem = 1 mkmem = 10
mpw = 376 nfft = 27000 nkpt = 10
PAW method is used; the additional fine FFT grid is defined by:
mgfftf= 40 nfftf = 64000
================================================================================
P This job should need less than 41.508 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 2.985 Mbytes ; DEN or POT disk file : 0.979 Mbytes.
================================================================================
DATASET 3 : space group R-3 m (#166); Bravais hR (rhombohedral)
================================================================================
Values of the parameters that define the memory need for DATASET 3.
intxc = 0 ionmov = 0 iscf = 17 lmnmax = 18
lnmax = 6 mgfft = 30 mpssoang = 3 mqgrid = 3001
natom = 4 nloc_mem = 2 nspden = 2 nspinor = 1
nsppol = 2 nsym = 12 n1xccc = 1 ntypat = 3
occopt = 0 xclevel = 2
- mband = 26 mffmem = 1 mkmem = 10
mpw = 376 nfft = 27000 nkpt = 10
PAW method is used; the additional fine FFT grid is defined by:
mgfftf= 40 nfftf = 64000
================================================================================
P This job should need less than 41.508 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 2.985 Mbytes ; DEN or POT disk file : 0.979 Mbytes.
================================================================================
DATASET 4 : space group R-3 m (#166); Bravais hR (rhombohedral)
================================================================================
Values of the parameters that define the memory need for DATASET 4.
intxc = 0 ionmov = 0 iscf = 17 lmnmax = 18
lnmax = 6 mgfft = 30 mpssoang = 3 mqgrid = 3001
natom = 4 nloc_mem = 2 nspden = 2 nspinor = 1
nsppol = 2 nsym = 12 n1xccc = 1 ntypat = 3
occopt = 0 xclevel = 2
- mband = 26 mffmem = 1 mkmem = 10
mpw = 376 nfft = 27000 nkpt = 10
PAW method is used; the additional fine FFT grid is defined by:
mgfftf= 40 nfftf = 64000
================================================================================
P This job should need less than 41.508 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 2.985 Mbytes ; DEN or POT disk file : 0.979 Mbytes.
================================================================================
DATASET 5 : space group R-3 m (#166); Bravais hR (rhombohedral)
================================================================================
Values of the parameters that define the memory need for DATASET 5.
intxc = 0 ionmov = 0 iscf = 17 lmnmax = 18
lnmax = 6 mgfft = 30 mpssoang = 3 mqgrid = 3001
natom = 4 nloc_mem = 2 nspden = 2 nspinor = 1
nsppol = 2 nsym = 12 n1xccc = 1 ntypat = 3
occopt = 0 xclevel = 2
- mband = 26 mffmem = 1 mkmem = 10
mpw = 376 nfft = 27000 nkpt = 10
PAW method is used; the additional fine FFT grid is defined by:
mgfftf= 40 nfftf = 64000
================================================================================
P This job should need less than 41.508 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 2.985 Mbytes ; DEN or POT disk file : 0.979 Mbytes.
================================================================================
--------------------------------------------------------------------------------
------------- Echo of variables that govern the present computation ------------
--------------------------------------------------------------------------------
-
- outvars: echo of selected default values
- iomode0 = 0 , fftalg0 =512 , wfoptalg0 = 10
-
- outvars: echo of global parameters not present in the input file
- max_nthreads = 0
-
-outvars: echo values of preprocessed input variables --------
acell 7.8800000000E+00 7.8800000000E+00 7.8800000000E+00 Bohr
amu 5.86900000E+01 5.86900000E+01 1.59994000E+01
atvshift1 -0.00551 -0.00551 -0.00551 -0.00551 -0.00551
-0.00551 -0.00551 -0.00551 -0.00551 -0.00551
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
atvshift2 -0.00367 -0.00367 -0.00367 -0.00367 -0.00367
-0.00367 -0.00367 -0.00367 -0.00367 -0.00367
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
atvshift3 -0.00184 -0.00184 -0.00184 -0.00184 -0.00184
-0.00184 -0.00184 -0.00184 -0.00184 -0.00184
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
atvshift4 0.00184 0.00184 0.00184 0.00184 0.00184
0.00184 0.00184 0.00184 0.00184 0.00184
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
atvshift5 0.00367 0.00367 0.00367 0.00367 0.00367
0.00367 0.00367 0.00367 0.00367 0.00367
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
chkprim 0
chksymbreak 0
diemix 4.50000000E-01
dmatpuopt 3
ecut 1.00000000E+01 Hartree
- fftalg 512
ixc -101130
jdtset 1 2 3 4 5
kpt 1.25000000E-01 1.25000000E-01 1.25000000E-01
3.75000000E-01 1.25000000E-01 1.25000000E-01
-3.75000000E-01 1.25000000E-01 1.25000000E-01
-1.25000000E-01 1.25000000E-01 1.25000000E-01
3.75000000E-01 3.75000000E-01 1.25000000E-01
-3.75000000E-01 3.75000000E-01 1.25000000E-01
-1.25000000E-01 3.75000000E-01 1.25000000E-01
-3.75000000E-01 -3.75000000E-01 1.25000000E-01
3.75000000E-01 3.75000000E-01 3.75000000E-01
-3.75000000E-01 3.75000000E-01 3.75000000E-01
kptrlatt 4 0 0 0 4 0 0 0 4
kptrlen 2.22880057E+01
lpawu 2 2 1
macro_uj 1
P mkmem 10
natom 4
nband 26
natvshift 5
ndtset 5
ngfft 30 30 30
ngfftdg 40 40 40
nkpt 10
nspden 2
nsppol 2
nsym 12
ntypat 3
occ 1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
0.000000 0.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
0.000000 0.000000
occopt 0
pawecutdg 2.00000000E+01 Hartree
pawujv1 -5.5123983263E-03 Hartree
pawujv2 -3.6749322176E-03 Hartree
pawujv3 -1.8374661088E-03 Hartree
pawujv4 1.8374661088E-03 Hartree
pawujv5 3.6749322176E-03 Hartree
prtden 0
prtebands 0
prteig 0
prtgsr 0
prtwf 0
rprim 5.0000000000E-01 5.0000000000E-01 1.0000000000E+00
1.0000000000E+00 5.0000000000E-01 5.0000000000E-01
5.0000000000E-01 1.0000000000E+00 5.0000000000E-01
shiftk 5.00000000E-01 5.00000000E-01 5.00000000E-01
spgroup 166
spinat 0.0000000000E+00 0.0000000000E+00 3.0000000000E+00
0.0000000000E+00 0.0000000000E+00 -3.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
tolvrs 1.00000000E-09
typat 1 2 3 3
usepawu 1
useylm 1
wtk 0.03125 0.09375 0.09375 0.09375 0.09375 0.18750
0.18750 0.09375 0.03125 0.09375
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
4.1699164191E+00 4.1699164191E+00 4.1699164191E+00
2.0849582095E+00 2.0849582095E+00 2.0849582095E+00
6.2548746286E+00 6.2548746286E+00 6.2548746286E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
7.8800000000E+00 7.8800000000E+00 7.8800000000E+00
3.9400000000E+00 3.9400000000E+00 3.9400000000E+00
1.1820000000E+01 1.1820000000E+01 1.1820000000E+01
xred 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 5.0000000000E-01
2.5000000000E-01 2.5000000000E-01 2.5000000000E-01
7.5000000000E-01 7.5000000000E-01 7.5000000000E-01
znucl 28.00000 28.00000 8.00000
================================================================================
chkinp: Checking input parameters for consistency, jdtset= 1.
chkinp: Checking input parameters for consistency, jdtset= 2.
chkinp: Checking input parameters for consistency, jdtset= 3.
chkinp: Checking input parameters for consistency, jdtset= 4.
chkinp: Checking input parameters for consistency, jdtset= 5.
================================================================================
== DATASET 1 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 1, }
dimensions: {natom: 4, nkpt: 10, mband: 26, nsppol: 2, nspinor: 1, nspden: 2, mpw: 376, }
cutoff_energies: {ecut: 10.0, pawecutdg: 20.0, }
electrons: {nelect: 4.80000000E+01, charge: 0.00000000E+00, occopt: 0.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 0, ionmov: 0, optcell: 0, iscf: 17, paral_kgb: 0, }
...
mkfilename: getwfk from: ./tlruj_1.o_WFK
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 3.9400000 3.9400000 7.8800000 G(1)= -0.0634518 -0.0634518 0.1903553
R(2)= 7.8800000 3.9400000 3.9400000 G(2)= 0.1903553 -0.0634518 -0.0634518
R(3)= 3.9400000 7.8800000 3.9400000 G(3)= -0.0634518 0.1903553 -0.0634518
Unit cell volume ucvol= 2.4465194E+02 bohr^3
Angles (23,13,12)= 3.35573098E+01 3.35573098E+01 3.35573098E+01 degrees
Coarse grid specifications (used for wave-functions):
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 30 30 30
ecut(hartree)= 10.000 => boxcut(ratio)= 2.18911
Fine grid specifications (used for densities):
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 40 40 40
ecut(hartree)= 20.000 => boxcut(ratio)= 2.06006
--- Pseudopotential description ------------------------------------------------
- pspini: atom type 1 psp file is /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/Ni.xml
- pspatm: opening atomic psp file /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/Ni.xml
- pspatm : Reading pseudopotential header in XML form from /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/Ni.xml
Pseudopotential format is: paw10
basis_size (lnmax)= 6 (lmn_size= 18), orbitals= 0 0 1 1 2 2
Spheres core radius: rc_sph= 1.81432190
1 radial meshes are used:
- mesh 1: r(i)=AA*[exp(BB*(i-1))-1], size=1500 , AA= 0.29784E-03 BB= 0.83395E-02
Shapefunction is SIN type: shapef(r)=[sin(pi*r/rshp)/(pi*r/rshp)]**2
Radius for shape functions = 1.60094920
mmax= 1500
Radial grid used for partial waves is grid 1
Radial grid used for projectors is grid 1
Radial grid used for (t)core density is grid 1
Radial grid used for Vloc is grid 1
Radial grid used for pseudo valence density is grid 1
Mesh size for Vloc has been set to 1333 to avoid numerical noise.
Compensation charge density is not taken into account in XC energy/potential
pspatm: atomic psp has been read and splines computed
- pspini: atom type 2 psp file is /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/Ni.xml
- pspatm: opening atomic psp file /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/Ni.xml
- pspatm : Reading pseudopotential header in XML form from /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/Ni.xml
Pseudopotential format is: paw10
basis_size (lnmax)= 6 (lmn_size= 18), orbitals= 0 0 1 1 2 2
Spheres core radius: rc_sph= 1.81432190
1 radial meshes are used:
- mesh 1: r(i)=AA*[exp(BB*(i-1))-1], size=1500 , AA= 0.29784E-03 BB= 0.83395E-02
Shapefunction is SIN type: shapef(r)=[sin(pi*r/rshp)/(pi*r/rshp)]**2
Radius for shape functions = 1.60094920
mmax= 1500
Radial grid used for partial waves is grid 1
Radial grid used for projectors is grid 1
Radial grid used for (t)core density is grid 1
Radial grid used for Vloc is grid 1
Radial grid used for pseudo valence density is grid 1
Mesh size for Vloc has been set to 1333 to avoid numerical noise.
Compensation charge density is not taken into account in XC energy/potential
pspatm: atomic psp has been read and splines computed
- pspini: atom type 3 psp file is /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/O.xml
- pspatm: opening atomic psp file /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/O.xml
- pspatm : Reading pseudopotential header in XML form from /home/buildbot/ABINIT3/eos_gnu_13.2_mpich/baguetl_fix-nhat-fft/tests/Pspdir/Psdj_paw_pbe_std/O.xml
Pseudopotential format is: paw10
basis_size (lnmax)= 4 (lmn_size= 8), orbitals= 0 0 1 1
Spheres core radius: rc_sph= 1.41465230
1 radial meshes are used:
- mesh 1: r(i)=AA*[exp(BB*(i-1))-1], size=2001 , AA= 0.72565E-03 BB= 0.58052E-02
Shapefunction is SIN type: shapef(r)=[sin(pi*r/rshp)/(pi*r/rshp)]**2
Radius for shape functions = 1.20231231
mmax= 2001
Radial grid used for partial waves is grid 1
Radial grid used for projectors is grid 1
Radial grid used for (t)core density is grid 1
Radial grid used for Vloc is grid 1
Radial grid used for pseudo valence density is grid 1
Mesh size for Vloc has been set to 1762 to avoid numerical noise.
Compensation charge density is not taken into account in XC energy/potential
pspatm: atomic psp has been read and splines computed
2.55203824E+03 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file ./tlruj_1.o_WFK
_setup2: Arith. and geom. avg. npw (full set) are 368.875 368.830
******************************************
DFT+U Method used: FLL
******************************************
================================================================================
--- !BeginCycle
iteration_state: {dtset: 1, }
solver: {iscf: 17, nstep: 30, nline: 4, wfoptalg: 10, }
tolerances: {tolvrs: 1.00E-09, }
...
iter Etot(hartree) deltaE(h) residm nres2 difmagsph maxmagsph
ETOT 1 -363.75876618626 -3.638E+02 1.102E-07 4.793E-01 1.354E-01 1.354E-01
ETOT 2 -363.76534979445 -6.584E-03 1.367E-06 1.057E+01 7.889E-02 7.194E-02
ETOT 3 -363.75845927752 6.891E-03 4.526E-04 2.860E-02 9.334E-02 1.498E-01
ETOT 4 -363.75846308828 -3.811E-06 1.401E-06 6.914E-04 1.627E-03 1.514E-01
ETOT 5 -363.75846321641 -1.281E-07 6.874E-07 3.264E-04 3.514E-04 1.518E-01
ETOT 6 -363.75846372876 -5.124E-07 1.876E-07 5.345E-04 5.811E-04 1.524E-01
ETOT 7 -363.75846369515 3.361E-08 5.981E-08 6.175E-05 4.975E-04 1.529E-01
ETOT 8 -363.75846382422 -1.291E-07 3.338E-08 2.851E-05 5.767E-04 1.534E-01
ETOT 9 -363.75846386761 -4.339E-08 1.489E-08 2.469E-05 3.138E-04 1.538E-01
ETOT 10 -363.75846393937 -7.176E-08 2.713E-08 1.216E-05 1.096E-03 1.549E-01
ETOT 11 -363.75846396570 -2.633E-08 6.303E-09 1.492E-06 7.186E-04 1.556E-01
ETOT 12 -363.75846397581 -1.010E-08 4.372E-09 4.494E-07 2.040E-04 1.558E-01
ETOT 13 -363.75846398647 -1.067E-08 1.512E-09 1.079E-07 1.406E-04 1.559E-01
ETOT 14 -363.75846398741 -9.416E-10 9.336E-10 1.567E-08 4.952E-05 1.560E-01
ETOT 15 -363.75846398774 -3.321E-10 2.791E-10 2.745E-09 2.491E-05 1.560E-01
ETOT 16 -363.75846398781 -6.241E-11 1.390E-10 1.585E-09 5.258E-06 1.560E-01
ETOT 17 -363.75846398781 -7.049E-12 4.826E-11 2.596E-09 1.478E-06 1.560E-01
ETOT 18 -363.75846398782 -2.785E-12 1.902E-11 3.909E-11 2.915E-06 1.560E-01
At SCF step 18 nres2 = 3.91E-11 < tolvrs= 1.00E-09 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42483938E-01 sigma(3 2)= -4.85585376E-04
sigma(2 2)= 1.42483938E-01 sigma(3 1)= -4.85585376E-04
sigma(3 3)= 1.42483938E-01 sigma(2 1)= -4.85585376E-04
--- !ResultsGS
iteration_state: {dtset: 1, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.9400000, 3.9400000, 7.8800000, ]
- [ 7.8800000, 3.9400000, 3.9400000, ]
- [ 3.9400000, 7.8800000, 3.9400000, ]
lattice_lengths: [ 9.65099, 9.65099, 9.65099, ]
lattice_angles: [ 33.557, 33.557, 33.557, ] # degrees, (23, 13, 12)
lattice_volume: 2.4465194E+02
convergence: {deltae: -2.785E-12, res2: 3.909E-11, residm: 1.902E-11, diffor: null, }
etotal : -3.63758464E+02
entropy : 0.00000000E+00
fermie : 4.06886672E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.42483938E-01, -4.85585376E-04, -4.85585376E-04, ]
- [ -4.85585376E-04, 1.42483938E-01, -4.85585376E-04, ]
- [ -4.85585376E-04, -4.85585376E-04, 1.42483938E-01, ]
pressure_GPa: -4.1920E+03
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Ni]
- [ 5.0000E-01, 5.0000E-01, 5.0000E-01, Ni]
- [ 2.5000E-01, 2.5000E-01, 2.5000E-01, O]
- [ 7.5000E-01, 7.5000E-01, 7.5000E-01, O]
cartesian_forces: # hartree/bohr
- [ 5.42101086E-20, -6.77626358E-20, -5.42101086E-20, ]
- [ 5.42101086E-20, -6.77626358E-20, -5.42101086E-20, ]
- [ -3.84191243E-04, -3.84191243E-04, -3.84191243E-04, ]
- [ 3.84191243E-04, 3.84191243E-04, 3.84191243E-04, ]
force_length_stats: {min: 1.02319336E-19, max: 6.65438753E-04, mean: 3.32719376E-04, }
...
Integrated electronic and magnetization densities in atomic spheres:
------------------------------------------------------------------------------------------------------------
Radius=ratsph(iatom), smearing ratsm= 0.0000. Diff(up-dn)=approximate z local magnetic moment.
Atom Radius up_density dn_density Total(up+dn) Diff(up-dn)
1 Ni 1.81432 8.027804 7.872077 15.899880 0.155727
2 Ni 1.81432 7.866218 8.022224 15.888442 -0.156007
3 O 1.41465 2.292181 2.291988 4.584169 0.000192
4 O 1.41465 2.292181 2.291988 4.584169 0.000192
------------------------------------------------------------------------------------------------------------
Sum: 20.478383 20.478278 40.956660 0.000105
Total magnetization (from the atomic spheres): 0.000105
Total magnetization (exact up - dn): -0.000000
PAW TEST:
==== Compensation charge inside spheres ============
The following values must be close to each other ...
Compensation charge over spherical meshes = 12.721410688917075
Compensation charge over fine fft grid = 12.721698036393011
==== Results concerning PAW augmentation regions ====
Total pseudopotential strength Dij (hartree):
Atom # 1 - Spin component 1
-2.32864 0.37495 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00170 -0.00170 0.00000 -0.00170 ...
0.37495 0.04455 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00034 0.00034 0.00000 0.00034 ...
0.00000 0.00000 -2.24018 -0.00125 -0.00125 0.82007 0.00076 0.00076 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00125 -2.24018 -0.00125 0.00076 0.82007 0.00076 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00125 -0.00125 -2.24018 0.00076 0.00076 0.82007 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.82007 0.00076 0.00076 0.37153 -0.00046 -0.00046 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00076 0.82007 0.00076 -0.00046 0.37153 -0.00046 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00076 0.00076 0.82007 -0.00046 -0.00046 0.37153 0.00000 0.00000 0.00000 0.00000 ...
-0.00170 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.91366 -0.00101 0.00093 -0.00101 ...
-0.00170 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00101 -0.91366 -0.00047 -0.00101 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00093 -0.00047 -0.91493 -0.00047 ...
-0.00170 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00101 -0.00101 -0.00047 -0.91366 ...
... only 12 components have been written...
Atom # 1 - Spin component 2
-2.32487 0.37426 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00223 -0.00223 0.00000 -0.00223 ...
0.37426 0.04466 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00044 0.00044 0.00000 0.00044 ...
0.00000 0.00000 -2.23702 -0.00165 -0.00165 0.81830 0.00099 0.00099 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00165 -2.23702 -0.00165 0.00099 0.81830 0.00099 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00165 -0.00165 -2.23702 0.00099 0.00099 0.81830 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.81830 0.00099 0.00099 0.37258 -0.00060 -0.00060 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00099 0.81830 0.00099 -0.00060 0.37258 -0.00060 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00099 0.00099 0.81830 -0.00060 -0.00060 0.37258 0.00000 0.00000 0.00000 0.00000 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.91093 -0.00114 0.00137 -0.00114 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00114 -0.91093 -0.00068 -0.00114 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00137 -0.00068 -0.91201 -0.00068 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00114 -0.00114 -0.00068 -0.91093 ...
... only 12 components have been written...
Atom # 4 - Spin component 1
0.61343 -1.75573 0.00001 0.00001 0.00001 -0.00004 -0.00004 -0.00004
-1.75573 5.08565 -0.00002 -0.00002 -0.00002 0.00010 0.00010 0.00010
0.00001 -0.00002 -0.29408 -0.00002 -0.00002 0.88876 0.00011 0.00011
0.00001 -0.00002 -0.00002 -0.29408 -0.00002 0.00011 0.88876 0.00011
0.00001 -0.00002 -0.00002 -0.00002 -0.29408 0.00011 0.00011 0.88876
-0.00004 0.00010 0.88876 0.00011 0.00011 0.11666 -0.00056 -0.00056
-0.00004 0.00010 0.00011 0.88876 0.00011 -0.00056 0.11666 -0.00056
-0.00004 0.00010 0.00011 0.00011 0.88876 -0.00056 -0.00056 0.11666
Atom # 4 - Spin component 2
0.61343 -1.75572 -0.00002 -0.00002 -0.00002 0.00011 0.00011 0.00011
-1.75572 5.08562 0.00004 0.00004 0.00004 -0.00029 -0.00029 -0.00029
-0.00002 0.00004 -0.29407 -0.00002 -0.00002 0.88874 0.00011 0.00011
-0.00002 0.00004 -0.00002 -0.29407 -0.00002 0.00011 0.88874 0.00011
-0.00002 0.00004 -0.00002 -0.00002 -0.29407 0.00011 0.00011 0.88874
0.00011 -0.00029 0.88874 0.00011 0.00011 0.11677 -0.00057 -0.00057
0.00011 -0.00029 0.00011 0.88874 0.00011 -0.00057 0.11677 -0.00057
0.00011 -0.00029 0.00011 0.00011 0.88874 -0.00057 -0.00057 0.11677
Augmentation waves occupancies Rhoij:
Atom # 1 - Spin component 1
0.97699 -0.16656 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00012 -0.00012 0.00000 -0.00012 ...
-0.16656 0.45536 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00320 -0.00320 0.00000 -0.00320 ...
0.00000 0.00000 1.06702 0.00002 0.00002 0.13588 0.00008 0.00008 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00002 1.06702 0.00002 0.00008 0.13588 0.00008 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00002 0.00002 1.06702 0.00008 0.00008 0.13588 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.13588 0.00008 0.00008 0.02463 0.00002 0.00002 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00008 0.13588 0.00008 0.00002 0.02463 0.00002 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00008 0.00008 0.13588 0.00002 0.00002 0.02463 0.00000 0.00000 0.00000 0.00000 ...
-0.00012 -0.00320 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 1.15589 -0.02172 0.02663 -0.02172 ...
-0.00012 -0.00320 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.02172 1.15589 -0.01331 -0.02172 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.02663 -0.01331 1.06102 -0.01331 ...
-0.00012 -0.00320 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.02172 -0.02172 -0.01331 1.15589 ...
... only 12 components have been written...
Atom # 1 - Spin component 2
0.97691 -0.16711 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00014 -0.00014 0.00000 -0.00014 ...
-0.16711 0.45290 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00182 -0.00182 0.00000 -0.00182 ...
0.00000 0.00000 1.06700 0.00003 0.00003 0.13578 0.00012 0.00012 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00003 1.06700 0.00003 0.00012 0.13578 0.00012 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00003 0.00003 1.06700 0.00012 0.00012 0.13578 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.13578 0.00012 0.00012 0.02454 0.00003 0.00003 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00012 0.13578 0.00012 0.00003 0.02454 0.00003 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00012 0.00012 0.13578 0.00003 0.00003 0.02454 0.00000 0.00000 0.00000 0.00000 ...
-0.00014 -0.00182 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 1.11188 -0.01376 0.00074 -0.01376 ...
-0.00014 -0.00182 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.01376 1.11188 -0.00037 -0.01376 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00074 -0.00037 1.01378 -0.00037 ...
-0.00014 -0.00182 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.01376 -0.01376 -0.00037 1.11188 ...
... only 12 components have been written...
Atom # 4 - Spin component 1
1.01156 0.01395 -0.00082 -0.00082 -0.00082 0.00003 0.00003 0.00003
0.01395 0.00119 0.00015 0.00015 0.00015 0.00000 0.00000 0.00000
-0.00082 0.00015 1.06817 -0.00323 -0.00323 0.05050 -0.00033 -0.00033
-0.00082 0.00015 -0.00323 1.06817 -0.00323 -0.00033 0.05050 -0.00033
-0.00082 0.00015 -0.00323 -0.00323 1.06817 -0.00033 -0.00033 0.05050
0.00003 0.00000 0.05050 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
0.00003 0.00000 -0.00033 0.05050 -0.00033 -0.00001 0.00287 -0.00001
0.00003 0.00000 -0.00033 -0.00033 0.05050 -0.00001 -0.00001 0.00287
Atom # 4 - Spin component 2
1.01152 0.01396 0.00053 0.00053 0.00053 -0.00003 -0.00003 -0.00003
0.01396 0.00119 -0.00007 -0.00007 -0.00007 0.00000 0.00000 0.00000
0.00053 -0.00007 1.06810 -0.00321 -0.00321 0.05050 -0.00033 -0.00033
0.00053 -0.00007 -0.00321 1.06810 -0.00321 -0.00033 0.05050 -0.00033
0.00053 -0.00007 -0.00321 -0.00321 1.06810 -0.00033 -0.00033 0.05050
-0.00003 0.00000 0.05050 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00003 0.00000 -0.00033 0.05050 -0.00033 -0.00001 0.00287 -0.00001
-0.00003 0.00000 -0.00033 -0.00033 0.05050 -0.00001 -0.00001 0.00287
"PAW+U" part of augmentation waves occupancies Rhoij:
Atom # 1 - L=2 ONLY - Spin component 1
1.15589 -0.02172 0.02663 -0.02172 0.00000 0.42978 -0.00881 0.01034 -0.00881 0.00000
-0.02172 1.15589 -0.01331 -0.02172 0.02306 -0.00881 0.42978 -0.00517 -0.00881 0.00895
0.02663 -0.01331 1.06102 -0.01331 0.00000 0.01204 -0.00602 0.39247 -0.00602 0.00000
-0.02172 -0.02172 -0.01331 1.15589 -0.02306 -0.00881 -0.00881 -0.00517 0.42978 -0.00895
0.00000 0.02306 0.00000 -0.02306 1.06102 0.00000 0.01043 0.00000 -0.01043 0.39247
0.42978 -0.00881 0.01204 -0.00881 0.00000 0.16436 -0.00361 0.00473 -0.00361 0.00000
-0.00881 0.42978 -0.00602 -0.00881 0.01043 -0.00361 0.16436 -0.00236 -0.00361 0.00410
0.01034 -0.00517 0.39247 -0.00517 0.00000 0.00473 -0.00236 0.15215 -0.00236 0.00000
-0.00881 -0.00881 -0.00602 0.42978 -0.01043 -0.00361 -0.00361 -0.00236 0.16436 -0.00410
0.00000 0.00895 0.00000 -0.00895 0.39247 0.00000 0.00410 0.00000 -0.00410 0.15215
Atom # 1 - L=2 ONLY - Spin component 2
1.11188 -0.01376 0.00074 -0.01376 0.00000 0.40969 -0.00539 -0.00014 -0.00539 0.00000
-0.01376 1.11188 -0.00037 -0.01376 0.00064 -0.00539 0.40969 0.00007 -0.00539 -0.00012
0.00074 -0.00037 1.01378 -0.00037 0.00000 -0.00033 0.00017 0.37453 0.00017 0.00000
-0.01376 -0.01376 -0.00037 1.11188 -0.00064 -0.00539 -0.00539 0.00007 0.40969 0.00012
0.00000 0.00064 0.00000 -0.00064 1.01378 0.00000 -0.00029 0.00000 0.00029 0.37453
0.40969 -0.00539 -0.00033 -0.00539 0.00000 0.15518 -0.00214 -0.00026 -0.00214 0.00000
-0.00539 0.40969 0.00017 -0.00539 -0.00029 -0.00214 0.15518 0.00013 -0.00214 -0.00023
-0.00014 0.00007 0.37453 0.00007 0.00000 -0.00026 0.00013 0.14529 0.00013 0.00000
-0.00539 -0.00539 0.00017 0.40969 0.00029 -0.00214 -0.00214 0.00013 0.15518 0.00023
0.00000 -0.00012 0.00000 0.00012 0.37453 0.00000 -0.00023 0.00000 0.00023 0.14529
Atom # 2 - L=2 ONLY - Spin component 1
1.11007 -0.01371 0.00055 -0.01371 0.00000 0.40876 -0.00537 -0.00026 -0.00537 0.00000
-0.01371 1.11007 -0.00028 -0.01371 0.00048 -0.00537 0.40876 0.00013 -0.00537 -0.00022
0.00055 -0.00028 1.01105 -0.00028 0.00000 -0.00050 0.00025 0.37338 0.00025 0.00000
-0.01371 -0.01371 -0.00028 1.11007 -0.00048 -0.00537 -0.00537 0.00013 0.40876 0.00022
0.00000 0.00048 0.00000 -0.00048 1.01105 0.00000 -0.00043 0.00000 0.00043 0.37338
0.40876 -0.00537 -0.00050 -0.00537 0.00000 0.15474 -0.00213 -0.00034 -0.00213 0.00000
-0.00537 0.40876 0.00025 -0.00537 -0.00043 -0.00213 0.15474 0.00017 -0.00213 -0.00030
-0.00026 0.00013 0.37338 0.00013 0.00000 -0.00034 0.00017 0.14484 0.00017 0.00000
-0.00537 -0.00537 0.00025 0.40876 0.00043 -0.00213 -0.00213 0.00017 0.15474 0.00030
0.00000 -0.00022 0.00000 0.00022 0.37338 0.00000 -0.00030 0.00000 0.00030 0.14484
Atom # 2 - L=2 ONLY - Spin component 2
1.15432 -0.02174 0.02644 -0.02174 0.00000 0.42901 -0.00882 0.01022 -0.00882 0.00000
-0.02174 1.15432 -0.01322 -0.02174 0.02290 -0.00882 0.42901 -0.00511 -0.00882 0.00885
0.02644 -0.01322 1.05818 -0.01322 0.00000 0.01190 -0.00595 0.39128 -0.00595 0.00000
-0.02174 -0.02174 -0.01322 1.15432 -0.02290 -0.00882 -0.00882 -0.00511 0.42901 -0.00885
0.00000 0.02290 0.00000 -0.02290 1.05818 0.00000 0.01031 0.00000 -0.01031 0.39128
0.42901 -0.00882 0.01190 -0.00882 0.00000 0.16402 -0.00361 0.00466 -0.00361 0.00000
-0.00882 0.42901 -0.00595 -0.00882 0.01031 -0.00361 0.16402 -0.00233 -0.00361 0.00404
0.01022 -0.00511 0.39128 -0.00511 0.00000 0.00466 -0.00233 0.15168 -0.00233 0.00000
-0.00882 -0.00882 -0.00595 0.42901 -0.01031 -0.00361 -0.00361 -0.00233 0.16402 -0.00404
0.00000 0.00885 0.00000 -0.00885 0.39128 0.00000 0.00404 0.00000 -0.00404 0.15168
Atom # 3 - L=1 ONLY - Spin component 1
1.06817 -0.00323 -0.00323 0.05050 -0.00033 -0.00033
-0.00323 1.06817 -0.00323 -0.00033 0.05050 -0.00033
-0.00323 -0.00323 1.06817 -0.00033 -0.00033 0.05050
0.05050 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05050 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05050 -0.00001 -0.00001 0.00287
Atom # 3 - L=1 ONLY - Spin component 2
1.06810 -0.00321 -0.00321 0.05050 -0.00033 -0.00033
-0.00321 1.06810 -0.00321 -0.00033 0.05050 -0.00033
-0.00321 -0.00321 1.06810 -0.00033 -0.00033 0.05050
0.05050 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05050 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05050 -0.00001 -0.00001 0.00287
Atom # 4 - L=1 ONLY - Spin component 1
1.06817 -0.00323 -0.00323 0.05050 -0.00033 -0.00033
-0.00323 1.06817 -0.00323 -0.00033 0.05050 -0.00033
-0.00323 -0.00323 1.06817 -0.00033 -0.00033 0.05050
0.05050 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05050 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05050 -0.00001 -0.00001 0.00287
Atom # 4 - L=1 ONLY - Spin component 2
1.06810 -0.00321 -0.00321 0.05050 -0.00033 -0.00033
-0.00321 1.06810 -0.00321 -0.00033 0.05050 -0.00033
-0.00321 -0.00321 1.06810 -0.00033 -0.00033 0.05050
0.05050 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05050 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05050 -0.00001 -0.00001 0.00287
---------- DFT+U DATA ---------------------------------------------------
====== For Atom 1, occupations for correlated orbitals. lpawu = 2
Atom 1. Occ. for lpawu and for spin 1 = 4.41365
Atom 1. Occ. for lpawu and for spin 2 = 4.23843
=> On atom 1, local Mag. for lpawu is -0.175220
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.91241 -0.01692 0.02063 -0.01692 0.00000
-0.01692 0.91241 -0.01032 -0.01692 0.01787
0.02063 -0.01032 0.83821 -0.01032 0.00000
-0.01692 -0.01692 -0.01032 0.91241 -0.01787
0.00000 0.01787 0.00000 -0.01787 0.83821
Occupation matrix for spin 2
0.87878 -0.01078 0.00073 -0.01078 0.00000
-0.01078 0.87878 -0.00037 -0.01078 0.00064
0.00073 -0.00037 0.80104 -0.00037 0.00000
-0.01078 -0.01078 -0.00037 0.87878 -0.00064
0.00000 0.00064 0.00000 -0.00064 0.80104
====== For Atom 2, occupations for correlated orbitals. lpawu = 2
Atom 2. Occ. for lpawu and for spin 1 = 4.23015
Atom 2. Occ. for lpawu and for spin 2 = 4.40570
=> On atom 2, local Mag. for lpawu is 0.175555
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.87743 -0.01074 0.00061 -0.01074 0.00000
-0.01074 0.87743 -0.00031 -0.01074 0.00053
0.00061 -0.00031 0.79893 -0.00031 0.00000
-0.01074 -0.01074 -0.00031 0.87743 -0.00053
0.00000 0.00053 0.00000 -0.00053 0.79893
Occupation matrix for spin 2
0.91123 -0.01694 0.02050 -0.01694 0.00000
-0.01694 0.91123 -0.01025 -0.01694 0.01775
0.02050 -0.01025 0.83601 -0.01025 0.00000
-0.01694 -0.01694 -0.01025 0.91123 -0.01775
0.00000 0.01775 0.00000 -0.01775 0.83601
====== For Atom 3, occupations for correlated orbitals. lpawu = 1
Atom 3. Occ. for lpawu and for spin 1 = 1.55460
Atom 3. Occ. for lpawu and for spin 2 = 1.55446
=> On atom 3, local Mag. for lpawu is -0.000140
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.51820 -0.00084 -0.00084
-0.00084 0.51820 -0.00084
-0.00084 -0.00084 0.51820
Occupation matrix for spin 2
0.51815 -0.00082 -0.00082
-0.00082 0.51815 -0.00082
-0.00082 -0.00082 0.51815
====== For Atom 4, occupations for correlated orbitals. lpawu = 1
Atom 4. Occ. for lpawu and for spin 1 = 1.55460
Atom 4. Occ. for lpawu and for spin 2 = 1.55446
=> On atom 4, local Mag. for lpawu is -0.000140
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.51820 -0.00084 -0.00084
-0.00084 0.51820 -0.00084
-0.00084 -0.00084 0.51820
Occupation matrix for spin 2
0.51815 -0.00082 -0.00082
-0.00082 0.51815 -0.00082
-0.00082 -0.00082 0.51815
---------- calculate U, (J) start ----------
All pawujat ok and equal to 1
macro_uj=1 and nspden=2: standard determination of Hubbard U-parameter
*********************************************************************
************************ Linear Response U ************************
Info printed for perturbed atom: 1
Perturbations Occupations
--------------- -----------------------------
alpha [eV] Unscreened Screened
--------------- -----------------------------
0.0000000000 8.6380182458 8.6380182458
-0.1500000000 8.6964981863 8.6520721987
Scalar response functions:
Chi0 [eV^-1]: -0.86637
Chi [eV^-1]: -0.09369
The scalar U from the two-point regression scheme is 9.51891 eV.
*********************************************************************
*********************************************************************
Note: For more reliable linear regressions of the response
matrices, it is advised that you have more than two points.
See the LRUJ protocol for more information.
URES ii nat r_max U(J)[eV] U_ASA[eV] U_inf[eV]
URES 1 1 0.00000 2.37973 1.91505 1.85249
URES 2 8 11.14400 8.34374 6.71448 6.49514
URES 3 27 12.45940 9.16681 7.37684 7.13586
URES 4 64 22.28800 9.37021 7.54052 7.29420
URES 5 125 24.28780 9.44277 7.59891 7.35068
URES 6 216 33.43200 9.47484 7.62472 7.37565
---------- calculate U, (J) end --------------
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 10.681E-14; max= 19.017E-12
reduced coordinates (array xred) for 4 atoms
0.000000000000 0.000000000000 0.000000000000
0.500000000000 0.500000000000 0.500000000000
0.250000000000 0.250000000000 0.250000000000
0.750000000000 0.750000000000 0.750000000000
rms dE/dt= 4.2814E-03; max dE/dt= 6.0549E-03; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
2 0.000000000000 0.000000000000 0.000000000000
3 0.006054853989 0.006054853989 0.006054853989
4 -0.006054853989 -0.006054853989 -0.006054853989
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
2 4.16991641908672 4.16991641908672 4.16991641908672
3 2.08495820954336 2.08495820954336 2.08495820954336
4 6.25487462863008 6.25487462863008 6.25487462863008
cartesian forces (hartree/bohr) at end:
1 0.00000000000000 -0.00000000000000 -0.00000000000000
2 0.00000000000000 -0.00000000000000 -0.00000000000000
3 -0.00038419124296 -0.00038419124296 -0.00038419124296
4 0.00038419124296 0.00038419124296 0.00038419124296
frms,max,avg= 2.7166423E-04 3.8419124E-04 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 0.00000000000000 -0.00000000000000 -0.00000000000000
2 0.00000000000000 -0.00000000000000 -0.00000000000000
3 -0.01975590803250 -0.01975590803250 -0.01975590803250
4 0.01975590803250 0.01975590803250 0.01975590803250
frms,max,avg= 1.3969537E-02 1.9755908E-02 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.880000000000 7.880000000000 7.880000000000 bohr
= 4.169916419087 4.169916419087 4.169916419087 angstroms
Fermi (or HOMO) energy (hartree) = 0.40689 Average Vxc (hartree)= -0.46897
Eigenvalues (hartree) for nkpt= 10 k points, SPIN UP:
kpt# 1, nband= 26, wtk= 0.03125, kpt= 0.1250 0.1250 0.1250 (reduced coord)
-4.13802 -4.13610 -2.36951 -2.33717 -2.33037 -2.33037 -2.30834 -2.30834
-0.40354 -0.35639 0.07010 0.21050 0.21050 0.22570 0.22570 0.25257
0.26128 0.26128 0.31826 0.31826 0.37388 0.38649 0.38649 0.39600
0.40070 0.43571
prteigrs : prtvol=0 or 1, do not print more k-points.
Eigenvalues (hartree) for nkpt= 10 k points, SPIN DOWN:
kpt# 1, nband= 26, wtk= 0.03125, kpt= 0.1250 0.1250 0.1250 (reduced coord)
-4.14324 -4.13088 -2.37027 -2.33643 -2.33217 -2.33217 -2.30654 -2.30654
-0.40354 -0.35638 0.07009 0.21051 0.21051 0.22570 0.22570 0.25258
0.26129 0.26129 0.31825 0.31825 0.37396 0.38650 0.38650 0.39597
0.40063 0.43568
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 1, }
comment : Components of total free energy in Hartree
kinetic : 8.94961346927132E+01
hartree : 8.65906815426754E+01
xc : -2.18505986223930E+01
Ewald energy : -2.39331554544165E+02
psp_core : 1.04313020493122E+01
local_psp : -2.68648474200428E+02
PAW spherical_terms : -2.03982622981769E+01
total_energy : -3.63710771380463E+02
total_energy_eV : -9.89707428185746E+03
...
--- !EnergyTermsDC
iteration_state : {dtset: 1, }
comment : '"Double-counting" decomposition of free energy'
band_energy : -4.00263798491403E+01
Ewald energy : -2.39331554544165E+02
psp_core : 1.04313020493122E+01
xc_dc : -8.00302998779472E+01
PAW spherical_terms : -1.48015317658759E+01
total_energy_dc : -3.63758463987817E+02
total_energy_dc_eV : -9.89837206381725E+03
...
--- !EnergyTermsWithPAWCore
iteration_state : {dtset: 1, }
comment : 'Components of total free energy, including PAW core contributions'
Total energy : -3.18179006382454E+03
Total energy DC : -3.18183775643190E+03
Total energy (eV) : -8.65809183803542E+04
Total energy DC (eV): -8.65822161623140E+04
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42483938E-01 sigma(3 2)= -4.85585376E-04
sigma(2 2)= 1.42483938E-01 sigma(3 1)= -4.85585376E-04
sigma(3 3)= 1.42483938E-01 sigma(2 1)= -4.85585376E-04
-Cartesian components of stress tensor (GPa) [Pressure= -4.1920E+03 GPa]
- sigma(1 1)= 4.19202218E+03 sigma(3 2)= -1.42864150E+01
- sigma(2 2)= 4.19202218E+03 sigma(3 1)= -1.42864150E+01
- sigma(3 3)= 4.19202218E+03 sigma(2 1)= -1.42864150E+01
================================================================================
== DATASET 2 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 2, }
dimensions: {natom: 4, nkpt: 10, mband: 26, nsppol: 2, nspinor: 1, nspden: 2, mpw: 376, }
cutoff_energies: {ecut: 10.0, pawecutdg: 20.0, }
electrons: {nelect: 4.80000000E+01, charge: 0.00000000E+00, occopt: 0.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 0, ionmov: 0, optcell: 0, iscf: 17, paral_kgb: 0, }
...
mkfilename: getwfk from: ./tlruj_1.o_WFK
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 3.9400000 3.9400000 7.8800000 G(1)= -0.0634518 -0.0634518 0.1903553
R(2)= 7.8800000 3.9400000 3.9400000 G(2)= 0.1903553 -0.0634518 -0.0634518
R(3)= 3.9400000 7.8800000 3.9400000 G(3)= -0.0634518 0.1903553 -0.0634518
Unit cell volume ucvol= 2.4465194E+02 bohr^3
Angles (23,13,12)= 3.35573098E+01 3.35573098E+01 3.35573098E+01 degrees
Coarse grid specifications (used for wave-functions):
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 30 30 30
ecut(hartree)= 10.000 => boxcut(ratio)= 2.18911
Fine grid specifications (used for densities):
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 40 40 40
ecut(hartree)= 20.000 => boxcut(ratio)= 2.06006
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file ./tlruj_1.o_WFK
_setup2: Arith. and geom. avg. npw (full set) are 368.875 368.830
******************************************
DFT+U Method used: FLL
******************************************
================================================================================
--- !BeginCycle
iteration_state: {dtset: 2, }
solver: {iscf: 17, nstep: 30, nline: 4, wfoptalg: 10, }
tolerances: {tolvrs: 1.00E-09, }
...
iter Etot(hartree) deltaE(h) residm nres2 difmagsph maxmagsph
ETOT 1 -363.74270119640 -3.637E+02 5.081E-08 1.911E-01 1.495E-01 1.495E-01
ETOT 2 -363.74528981457 -2.589E-03 5.358E-07 5.178E+00 8.012E-02 7.859E-02
ETOT 3 -363.74256595333 2.724E-03 1.722E-04 6.078E-03 8.394E-02 1.533E-01
ETOT 4 -363.74257000093 -4.048E-06 4.625E-07 1.327E-04 6.249E-04 1.540E-01
ETOT 5 -363.74257042722 -4.263E-07 2.689E-07 5.670E-04 2.840E-04 1.542E-01
ETOT 6 -363.74257027864 1.486E-07 1.144E-08 1.175E-04 2.498E-04 1.545E-01
ETOT 7 -363.74257030028 -2.164E-08 4.787E-09 4.089E-05 2.776E-04 1.548E-01
ETOT 8 -363.74257030949 -9.210E-09 1.397E-10 3.213E-05 9.508E-05 1.549E-01
ETOT 9 -363.74257032900 -1.950E-08 1.594E-09 1.635E-05 3.114E-04 1.552E-01
ETOT 10 -363.74257033749 -8.495E-09 1.390E-10 1.577E-05 1.386E-04 1.553E-01
ETOT 11 -363.74257034769 -1.019E-08 5.504E-09 1.591E-06 7.877E-04 1.561E-01
ETOT 12 -363.74257035386 -6.172E-09 3.838E-11 5.789E-07 7.563E-05 1.562E-01
ETOT 13 -363.74257035770 -3.841E-09 1.396E-10 1.316E-07 8.459E-05 1.563E-01
ETOT 14 -363.74257035980 -2.105E-09 6.476E-11 1.430E-08 7.525E-05 1.563E-01
ETOT 15 -363.74257036009 -2.885E-10 2.728E-11 1.283E-08 1.358E-05 1.563E-01
ETOT 16 -363.74257036015 -5.804E-11 1.118E-11 4.237E-09 7.338E-06 1.564E-01
ETOT 17 -363.74257036015 -1.307E-12 1.076E-12 4.346E-10 2.757E-06 1.564E-01
At SCF step 17 nres2 = 4.35E-10 < tolvrs= 1.00E-09 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42479676E-01 sigma(3 2)= -4.86299685E-04
sigma(2 2)= 1.42479676E-01 sigma(3 1)= -4.86299685E-04
sigma(3 3)= 1.42479676E-01 sigma(2 1)= -4.86299685E-04
--- !ResultsGS
iteration_state: {dtset: 2, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.9400000, 3.9400000, 7.8800000, ]
- [ 7.8800000, 3.9400000, 3.9400000, ]
- [ 3.9400000, 7.8800000, 3.9400000, ]
lattice_lengths: [ 9.65099, 9.65099, 9.65099, ]
lattice_angles: [ 33.557, 33.557, 33.557, ] # degrees, (23, 13, 12)
lattice_volume: 2.4465194E+02
convergence: {deltae: -1.307E-12, res2: 4.346E-10, residm: 1.076E-12, diffor: null, }
etotal : -3.63742570E+02
entropy : 0.00000000E+00
fermie : 4.07284737E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.42479676E-01, -4.86299685E-04, -4.86299685E-04, ]
- [ -4.86299685E-04, 1.42479676E-01, -4.86299685E-04, ]
- [ -4.86299685E-04, -4.86299685E-04, 1.42479676E-01, ]
pressure_GPa: -4.1919E+03
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Ni]
- [ 5.0000E-01, 5.0000E-01, 5.0000E-01, Ni]
- [ 2.5000E-01, 2.5000E-01, 2.5000E-01, O]
- [ 7.5000E-01, 7.5000E-01, 7.5000E-01, O]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, 2.71050543E-20, -5.42101086E-20, ]
- [ -0.00000000E+00, 2.71050543E-20, -5.42101086E-20, ]
- [ -2.57270305E-04, -2.57270305E-04, -2.57270305E-04, ]
- [ 2.57270305E-04, 2.57270305E-04, 2.57270305E-04, ]
force_length_stats: {min: 6.06087440E-20, max: 4.45605240E-04, mean: 2.22802620E-04, }
...
Integrated electronic and magnetization densities in atomic spheres:
------------------------------------------------------------------------------------------------------------
Radius=ratsph(iatom), smearing ratsm= 0.0000. Diff(up-dn)=approximate z local magnetic moment.
Atom Radius up_density dn_density Total(up+dn) Diff(up-dn)
1 Ni 1.81432 8.026377 7.870255 15.896632 0.156122
2 Ni 1.81432 7.866323 8.022678 15.889001 -0.156355
3 O 1.41465 2.292227 2.292090 4.584317 0.000137
4 O 1.41465 2.292227 2.292090 4.584317 0.000137
------------------------------------------------------------------------------------------------------------
Sum: 20.477154 20.477113 40.954267 0.000041
Total magnetization (from the atomic spheres): 0.000041
Total magnetization (exact up - dn): -0.000000
PAW TEST:
==== Compensation charge inside spheres ============
The following values must be close to each other ...
Compensation charge over spherical meshes = 12.720567589015431
Compensation charge over fine fft grid = 12.720854497892057
==== Results concerning PAW augmentation regions ====
Total pseudopotential strength Dij (hartree):
Atom # 1 - Spin component 1
-2.32959 0.37520 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00170 -0.00170 0.00000 -0.00170 ...
0.37520 0.04451 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00034 0.00034 0.00000 0.00034 ...
0.00000 0.00000 -2.24104 -0.00125 -0.00125 0.82074 0.00076 0.00076 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00125 -2.24104 -0.00125 0.00076 0.82074 0.00076 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00125 -0.00125 -2.24104 0.00076 0.00076 0.82074 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.82074 0.00076 0.00076 0.37110 -0.00046 -0.00046 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00076 0.82074 0.00076 -0.00046 0.37110 -0.00046 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00076 0.00076 0.82074 -0.00046 -0.00046 0.37110 0.00000 0.00000 0.00000 0.00000 ...
-0.00170 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.91299 -0.00101 0.00094 -0.00101 ...
-0.00170 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00101 -0.91299 -0.00047 -0.00101 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00094 -0.00047 -0.91427 -0.00047 ...
-0.00170 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00101 -0.00101 -0.00047 -0.91299 ...
... only 12 components have been written...
Atom # 1 - Spin component 2
-2.32581 0.37450 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00223 -0.00223 0.00000 -0.00223 ...
0.37450 0.04461 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00044 0.00044 0.00000 0.00044 ...
0.00000 0.00000 -2.23787 -0.00165 -0.00165 0.81896 0.00099 0.00099 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00165 -2.23787 -0.00165 0.00099 0.81896 0.00099 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00165 -0.00165 -2.23787 0.00099 0.00099 0.81896 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.81896 0.00099 0.00099 0.37216 -0.00060 -0.00060 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00099 0.81896 0.00099 -0.00060 0.37216 -0.00060 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00099 0.00099 0.81896 -0.00060 -0.00060 0.37216 0.00000 0.00000 0.00000 0.00000 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.91025 -0.00114 0.00137 -0.00114 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00114 -0.91025 -0.00068 -0.00114 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00137 -0.00068 -0.91133 -0.00068 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00114 -0.00114 -0.00068 -0.91025 ...
... only 12 components have been written...
Atom # 4 - Spin component 1
0.61342 -1.75567 0.00001 0.00001 0.00001 -0.00005 -0.00005 -0.00005
-1.75567 5.08546 -0.00002 -0.00002 -0.00002 0.00014 0.00014 0.00014
0.00001 -0.00002 -0.29405 -0.00002 -0.00002 0.88859 0.00011 0.00011
0.00001 -0.00002 -0.00002 -0.29405 -0.00002 0.00011 0.88859 0.00011
0.00001 -0.00002 -0.00002 -0.00002 -0.29405 0.00011 0.00011 0.88859
-0.00005 0.00014 0.88859 0.00011 0.00011 0.11755 -0.00056 -0.00056
-0.00005 0.00014 0.00011 0.88859 0.00011 -0.00056 0.11755 -0.00056
-0.00005 0.00014 0.00011 0.00011 0.88859 -0.00056 -0.00056 0.11755
Atom # 4 - Spin component 2
0.61341 -1.75566 -0.00002 -0.00002 -0.00002 0.00010 0.00010 0.00010
-1.75566 5.08543 0.00004 0.00004 0.00004 -0.00026 -0.00026 -0.00026
-0.00002 0.00004 -0.29405 -0.00002 -0.00002 0.88858 0.00011 0.00011
-0.00002 0.00004 -0.00002 -0.29405 -0.00002 0.00011 0.88858 0.00011
-0.00002 0.00004 -0.00002 -0.00002 -0.29405 0.00011 0.00011 0.88858
0.00010 -0.00026 0.88858 0.00011 0.00011 0.11763 -0.00057 -0.00057
0.00010 -0.00026 0.00011 0.88858 0.00011 -0.00057 0.11763 -0.00057
0.00010 -0.00026 0.00011 0.00011 0.88858 -0.00057 -0.00057 0.11763
Augmentation waves occupancies Rhoij:
Atom # 1 - Spin component 1
0.97696 -0.16669 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00013 -0.00013 0.00000 -0.00013 ...
-0.16669 0.45680 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00338 -0.00338 0.00000 -0.00338 ...
0.00000 0.00000 1.06703 0.00002 0.00002 0.13583 0.00008 0.00008 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00002 1.06703 0.00002 0.00008 0.13583 0.00008 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00002 0.00002 1.06703 0.00008 0.00008 0.13583 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.13583 0.00008 0.00008 0.02463 0.00002 0.00002 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00008 0.13583 0.00008 0.00002 0.02463 0.00002 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00008 0.00008 0.13583 0.00002 0.00002 0.02463 0.00000 0.00000 0.00000 0.00000 ...
-0.00013 -0.00338 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 1.15545 -0.02173 0.02663 -0.02173 ...
-0.00013 -0.00338 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.02173 1.15545 -0.01332 -0.02173 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.02663 -0.01332 1.06033 -0.01332 ...
-0.00013 -0.00338 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.02173 -0.02173 -0.01332 1.15545 ...
... only 12 components have been written...
Atom # 1 - Spin component 2
0.97689 -0.16722 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00013 -0.00013 0.00000 -0.00013 ...
-0.16722 0.45473 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00157 -0.00157 0.00000 -0.00157 ...
0.00000 0.00000 1.06700 0.00003 0.00003 0.13572 0.00012 0.00012 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00003 1.06700 0.00003 0.00012 0.13572 0.00012 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00003 0.00003 1.06700 0.00012 0.00012 0.13572 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.13572 0.00012 0.00012 0.02455 0.00003 0.00003 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00012 0.13572 0.00012 0.00003 0.02455 0.00003 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00012 0.00012 0.13572 0.00003 0.00003 0.02455 0.00000 0.00000 0.00000 0.00000 ...
-0.00013 -0.00157 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 1.11131 -0.01381 0.00070 -0.01381 ...
-0.00013 -0.00157 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.01381 1.11131 -0.00035 -0.01381 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00070 -0.00035 1.01297 -0.00035 ...
-0.00013 -0.00157 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.01381 -0.01381 -0.00035 1.11131 ...
... only 12 components have been written...
Atom # 4 - Spin component 1
1.01156 0.01395 -0.00079 -0.00079 -0.00079 0.00003 0.00003 0.00003
0.01395 0.00119 0.00014 0.00014 0.00014 0.00000 0.00000 0.00000
-0.00079 0.00014 1.06822 -0.00322 -0.00322 0.05051 -0.00033 -0.00033
-0.00079 0.00014 -0.00322 1.06822 -0.00322 -0.00033 0.05051 -0.00033
-0.00079 0.00014 -0.00322 -0.00322 1.06822 -0.00033 -0.00033 0.05051
0.00003 0.00000 0.05051 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
0.00003 0.00000 -0.00033 0.05051 -0.00033 -0.00001 0.00287 -0.00001
0.00003 0.00000 -0.00033 -0.00033 0.05051 -0.00001 -0.00001 0.00287
Atom # 4 - Spin component 2
1.01153 0.01396 0.00060 0.00060 0.00060 -0.00003 -0.00003 -0.00003
0.01396 0.00119 -0.00009 -0.00009 -0.00009 0.00000 0.00000 0.00000
0.00060 -0.00009 1.06817 -0.00321 -0.00321 0.05051 -0.00033 -0.00033
0.00060 -0.00009 -0.00321 1.06817 -0.00321 -0.00033 0.05051 -0.00033
0.00060 -0.00009 -0.00321 -0.00321 1.06817 -0.00033 -0.00033 0.05051
-0.00003 0.00000 0.05051 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00003 0.00000 -0.00033 0.05051 -0.00033 -0.00001 0.00287 -0.00001
-0.00003 0.00000 -0.00033 -0.00033 0.05051 -0.00001 -0.00001 0.00287
"PAW+U" part of augmentation waves occupancies Rhoij:
Atom # 1 - L=2 ONLY - Spin component 1
1.15545 -0.02173 0.02663 -0.02173 0.00000 0.42958 -0.00882 0.01033 -0.00882 0.00000
-0.02173 1.15545 -0.01332 -0.02173 0.02306 -0.00882 0.42958 -0.00517 -0.00882 0.00895
0.02663 -0.01332 1.06033 -0.01332 0.00000 0.01204 -0.00602 0.39217 -0.00602 0.00000
-0.02173 -0.02173 -0.01332 1.15545 -0.02306 -0.00882 -0.00882 -0.00517 0.42958 -0.00895
0.00000 0.02306 0.00000 -0.02306 1.06033 0.00000 0.01042 0.00000 -0.01042 0.39217
0.42958 -0.00882 0.01204 -0.00882 0.00000 0.16428 -0.00361 0.00473 -0.00361 0.00000
-0.00882 0.42958 -0.00602 -0.00882 0.01042 -0.00361 0.16428 -0.00236 -0.00361 0.00409
0.01033 -0.00517 0.39217 -0.00517 0.00000 0.00473 -0.00236 0.15203 -0.00236 0.00000
-0.00882 -0.00882 -0.00602 0.42958 -0.01042 -0.00361 -0.00361 -0.00236 0.16428 -0.00409
0.00000 0.00895 0.00000 -0.00895 0.39217 0.00000 0.00409 0.00000 -0.00409 0.15203
Atom # 1 - L=2 ONLY - Spin component 2
1.11131 -0.01381 0.00070 -0.01381 0.00000 0.40942 -0.00542 -0.00016 -0.00542 0.00000
-0.01381 1.11131 -0.00035 -0.01381 0.00061 -0.00542 0.40942 0.00008 -0.00542 -0.00014
0.00070 -0.00035 1.01297 -0.00035 0.00000 -0.00036 0.00018 0.37419 0.00018 0.00000
-0.01381 -0.01381 -0.00035 1.11131 -0.00061 -0.00542 -0.00542 0.00008 0.40942 0.00014
0.00000 0.00061 0.00000 -0.00061 1.01297 0.00000 -0.00031 0.00000 0.00031 0.37419
0.40942 -0.00542 -0.00036 -0.00542 0.00000 0.15506 -0.00215 -0.00028 -0.00215 0.00000
-0.00542 0.40942 0.00018 -0.00542 -0.00031 -0.00215 0.15506 0.00014 -0.00215 -0.00024
-0.00016 0.00008 0.37419 0.00008 0.00000 -0.00028 0.00014 0.14515 0.00014 0.00000
-0.00542 -0.00542 0.00018 0.40942 0.00031 -0.00215 -0.00215 0.00014 0.15506 0.00024
0.00000 -0.00014 0.00000 0.00014 0.37419 0.00000 -0.00024 0.00000 0.00024 0.14515
Atom # 2 - L=2 ONLY - Spin component 1
1.11008 -0.01381 0.00058 -0.01381 0.00000 0.40879 -0.00541 -0.00024 -0.00541 0.00000
-0.01381 1.11008 -0.00029 -0.01381 0.00050 -0.00541 0.40879 0.00012 -0.00541 -0.00021
0.00058 -0.00029 1.01115 -0.00029 0.00000 -0.00047 0.00024 0.37342 0.00024 0.00000
-0.01381 -0.01381 -0.00029 1.11008 -0.00050 -0.00541 -0.00541 0.00012 0.40879 0.00021
0.00000 0.00050 0.00000 -0.00050 1.01115 0.00000 -0.00041 0.00000 0.00041 0.37342
0.40879 -0.00541 -0.00047 -0.00541 0.00000 0.15476 -0.00215 -0.00033 -0.00215 0.00000
-0.00541 0.40879 0.00024 -0.00541 -0.00041 -0.00215 0.15476 0.00017 -0.00215 -0.00029
-0.00024 0.00012 0.37342 0.00012 0.00000 -0.00033 0.00017 0.14486 0.00017 0.00000
-0.00541 -0.00541 0.00024 0.40879 0.00041 -0.00215 -0.00215 0.00017 0.15476 0.00029
0.00000 -0.00021 0.00000 0.00021 0.37342 0.00000 -0.00029 0.00000 0.00029 0.14486
Atom # 2 - L=2 ONLY - Spin component 2
1.15441 -0.02174 0.02651 -0.02174 0.00000 0.42907 -0.00883 0.01026 -0.00883 0.00000
-0.02174 1.15441 -0.01325 -0.02174 0.02296 -0.00883 0.42907 -0.00513 -0.00883 0.00888
0.02651 -0.01325 1.05844 -0.01325 0.00000 0.01195 -0.00597 0.39138 -0.00597 0.00000
-0.02174 -0.02174 -0.01325 1.15441 -0.02296 -0.00883 -0.00883 -0.00513 0.42907 -0.00888
0.00000 0.02296 0.00000 -0.02296 1.05844 0.00000 0.01035 0.00000 -0.01035 0.39138
0.42907 -0.00883 0.01195 -0.00883 0.00000 0.16405 -0.00361 0.00468 -0.00361 0.00000
-0.00883 0.42907 -0.00597 -0.00883 0.01035 -0.00361 0.16405 -0.00234 -0.00361 0.00405
0.01026 -0.00513 0.39138 -0.00513 0.00000 0.00468 -0.00234 0.15172 -0.00234 0.00000
-0.00883 -0.00883 -0.00597 0.42907 -0.01035 -0.00361 -0.00361 -0.00234 0.16405 -0.00405
0.00000 0.00888 0.00000 -0.00888 0.39138 0.00000 0.00405 0.00000 -0.00405 0.15172
Atom # 3 - L=1 ONLY - Spin component 1
1.06822 -0.00322 -0.00322 0.05051 -0.00033 -0.00033
-0.00322 1.06822 -0.00322 -0.00033 0.05051 -0.00033
-0.00322 -0.00322 1.06822 -0.00033 -0.00033 0.05051
0.05051 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05051 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05051 -0.00001 -0.00001 0.00287
Atom # 3 - L=1 ONLY - Spin component 2
1.06817 -0.00321 -0.00321 0.05051 -0.00033 -0.00033
-0.00321 1.06817 -0.00321 -0.00033 0.05051 -0.00033
-0.00321 -0.00321 1.06817 -0.00033 -0.00033 0.05051
0.05051 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05051 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05051 -0.00001 -0.00001 0.00287
Atom # 4 - L=1 ONLY - Spin component 1
1.06822 -0.00322 -0.00322 0.05051 -0.00033 -0.00033
-0.00322 1.06822 -0.00322 -0.00033 0.05051 -0.00033
-0.00322 -0.00322 1.06822 -0.00033 -0.00033 0.05051
0.05051 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05051 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05051 -0.00001 -0.00001 0.00287
Atom # 4 - L=1 ONLY - Spin component 2
1.06817 -0.00321 -0.00321 0.05051 -0.00033 -0.00033
-0.00321 1.06817 -0.00321 -0.00033 0.05051 -0.00033
-0.00321 -0.00321 1.06817 -0.00033 -0.00033 0.05051
0.05051 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05051 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05051 -0.00001 -0.00001 0.00287
---------- DFT+U DATA ---------------------------------------------------
====== For Atom 1, occupations for correlated orbitals. lpawu = 2
Atom 1. Occ. for lpawu and for spin 1 = 4.41157
Atom 1. Occ. for lpawu and for spin 2 = 4.23586
=> On atom 1, local Mag. for lpawu is -0.175705
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.91207 -0.01693 0.02064 -0.01693 0.00000
-0.01693 0.91207 -0.01032 -0.01693 0.01787
0.02064 -0.01032 0.83768 -0.01032 0.00000
-0.01693 -0.01693 -0.01032 0.91207 -0.01787
0.00000 0.01787 0.00000 -0.01787 0.83768
Occupation matrix for spin 2
0.87835 -0.01082 0.00071 -0.01082 0.00000
-0.01082 0.87835 -0.00036 -0.01082 0.00062
0.00071 -0.00036 0.80041 -0.00036 0.00000
-0.01082 -0.01082 -0.00036 0.87835 -0.00062
0.00000 0.00062 0.00000 -0.00062 0.80041
====== For Atom 2, occupations for correlated orbitals. lpawu = 2
Atom 2. Occ. for lpawu and for spin 1 = 4.23030
Atom 2. Occ. for lpawu and for spin 2 = 4.40630
=> On atom 2, local Mag. for lpawu is 0.175995
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.87743 -0.01081 0.00063 -0.01081 0.00000
-0.01081 0.87743 -0.00031 -0.01081 0.00055
0.00063 -0.00031 0.79901 -0.00031 0.00000
-0.01081 -0.01081 -0.00031 0.87743 -0.00055
0.00000 0.00055 0.00000 -0.00055 0.79901
Occupation matrix for spin 2
0.91129 -0.01694 0.02055 -0.01694 0.00000
-0.01694 0.91129 -0.01028 -0.01694 0.01780
0.02055 -0.01028 0.83622 -0.01028 0.00000
-0.01694 -0.01694 -0.01028 0.91129 -0.01780
0.00000 0.01780 0.00000 -0.01780 0.83622
====== For Atom 3, occupations for correlated orbitals. lpawu = 1
Atom 3. Occ. for lpawu and for spin 1 = 1.55463
Atom 3. Occ. for lpawu and for spin 2 = 1.55453
=> On atom 3, local Mag. for lpawu is -0.000094
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.51821 -0.00083 -0.00083
-0.00083 0.51821 -0.00083
-0.00083 -0.00083 0.51821
Occupation matrix for spin 2
0.51818 -0.00082 -0.00082
-0.00082 0.51818 -0.00082
-0.00082 -0.00082 0.51818
====== For Atom 4, occupations for correlated orbitals. lpawu = 1
Atom 4. Occ. for lpawu and for spin 1 = 1.55463
Atom 4. Occ. for lpawu and for spin 2 = 1.55453
=> On atom 4, local Mag. for lpawu is -0.000094
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.51821 -0.00083 -0.00083
-0.00083 0.51821 -0.00083
-0.00083 -0.00083 0.51821
Occupation matrix for spin 2
0.51818 -0.00082 -0.00082
-0.00082 0.51818 -0.00082
-0.00082 -0.00082 0.51818
---------- calculate U, (J) start ----------
All pawujat ok and equal to 1
macro_uj=1 and nspden=2: standard determination of Hubbard U-parameter
*********************************************************************
************************ Linear Response U ************************
Info printed for perturbed atom: 1
Perturbations Occupations
--------------- -----------------------------
alpha [eV] Unscreened Screened
--------------- -----------------------------
0.0000000000 8.6380182458 8.6380182458
-0.1000000000 8.6747201546 8.6474287205
Scalar response functions:
Chi0 [eV^-1]: -0.81560
Chi [eV^-1]: -0.09410
The scalar U from the two-point regression scheme is 9.40036 eV.
*********************************************************************
*********************************************************************
Note: For more reliable linear regressions of the response
matrices, it is advised that you have more than two points.
See the LRUJ protocol for more information.
URES ii nat r_max U(J)[eV] U_ASA[eV] U_inf[eV]
URES 1 1 0.00000 2.35009 1.89120 1.82942
URES 2 8 11.14400 8.23982 6.63086 6.41425
URES 3 27 12.45940 9.05264 7.28497 7.04699
URES 4 64 22.28800 9.25352 7.44661 7.20336
URES 5 125 24.28780 9.32516 7.50427 7.25913
URES 6 216 33.43200 9.35684 7.52976 7.28379
---------- calculate U, (J) end --------------
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 53.823E-15; max= 10.764E-13
reduced coordinates (array xred) for 4 atoms
0.000000000000 0.000000000000 0.000000000000
0.500000000000 0.500000000000 0.500000000000
0.250000000000 0.250000000000 0.250000000000
0.750000000000 0.750000000000 0.750000000000
rms dE/dt= 2.8670E-03; max dE/dt= 4.0546E-03; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
2 0.000000000000 0.000000000000 0.000000000000
3 0.004054580014 0.004054580014 0.004054580014
4 -0.004054580014 -0.004054580014 -0.004054580014
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
2 4.16991641908672 4.16991641908672 4.16991641908672
3 2.08495820954336 2.08495820954336 2.08495820954336
4 6.25487462863008 6.25487462863008 6.25487462863008
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 0.00000000000000 -0.00000000000000
2 -0.00000000000000 0.00000000000000 -0.00000000000000
3 -0.00025727030544 -0.00025727030544 -0.00025727030544
4 0.00025727030544 0.00025727030544 0.00025727030544
frms,max,avg= 1.8191758E-04 2.5727031E-04 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 0.00000000000000 -0.00000000000000
2 -0.00000000000000 0.00000000000000 -0.00000000000000
3 -0.01322937101492 -0.01322937101492 -0.01322937101492
4 0.01322937101492 0.01322937101492 0.01322937101492
frms,max,avg= 9.3545780E-03 1.3229371E-02 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.880000000000 7.880000000000 7.880000000000 bohr
= 4.169916419087 4.169916419087 4.169916419087 angstroms
Fermi (or HOMO) energy (hartree) = 0.40728 Average Vxc (hartree)= -0.46901
Eigenvalues (hartree) for nkpt= 10 k points, SPIN UP:
kpt# 1, nband= 26, wtk= 0.03125, kpt= 0.1250 0.1250 0.1250 (reduced coord)
-4.13800 -4.13750 -2.37010 -2.33787 -2.33104 -2.33104 -2.30892 -2.30892
-0.40344 -0.35627 0.07016 0.21073 0.21073 0.22613 0.22613 0.25268
0.26145 0.26145 0.31868 0.31868 0.37432 0.38694 0.38694 0.39642
0.40071 0.43613
prteigrs : prtvol=0 or 1, do not print more k-points.
Eigenvalues (hartree) for nkpt= 10 k points, SPIN DOWN:
kpt# 1, nband= 26, wtk= 0.03125, kpt= 0.1250 0.1250 0.1250 (reduced coord)
-4.14275 -4.13276 -2.37061 -2.33737 -2.33227 -2.33227 -2.30770 -2.30770
-0.40343 -0.35626 0.07015 0.21074 0.21074 0.22613 0.22613 0.25269
0.26146 0.26146 0.31868 0.31868 0.37437 0.38695 0.38695 0.39640
0.40066 0.43610
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 2, }
comment : Components of total free energy in Hartree
kinetic : 8.94890712578726E+01
hartree : 8.65750590856252E+01
xc : -2.18498794965232E+01
Ewald energy : -2.39331554544165E+02
psp_core : 1.04313020493122E+01
local_psp : -2.68626248104743E+02
PAW spherical_terms : -2.03985418285443E+01
total_energy : -3.63710791581165E+02
total_energy_eV : -9.89707483154659E+03
...
--- !EnergyTermsDC
iteration_state : {dtset: 2, }
comment : '"Double-counting" decomposition of free energy'
band_energy : -4.00276152055839E+01
Ewald energy : -2.39331554544165E+02
psp_core : 1.04313020493122E+01
xc_dc : -8.00148744608467E+01
PAW spherical_terms : -1.47998281988672E+01
total_energy_dc : -3.63742570360151E+02
total_energy_dc_eV : -9.89793957617599E+03
...
--- !EnergyTermsWithPAWCore
iteration_state : {dtset: 2, }
comment : 'Components of total free energy, including PAW core contributions'
Total energy : -3.18179008402525E+03
Total energy DC : -3.18182186280423E+03
Total energy (eV) : -8.65809189300433E+04
Total energy DC (eV): -8.65817836746728E+04
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42479676E-01 sigma(3 2)= -4.86299685E-04
sigma(2 2)= 1.42479676E-01 sigma(3 1)= -4.86299685E-04
sigma(3 3)= 1.42479676E-01 sigma(2 1)= -4.86299685E-04
-Cartesian components of stress tensor (GPa) [Pressure= -4.1919E+03 GPa]
- sigma(1 1)= 4.19189678E+03 sigma(3 2)= -1.43074307E+01
- sigma(2 2)= 4.19189678E+03 sigma(3 1)= -1.43074307E+01
- sigma(3 3)= 4.19189678E+03 sigma(2 1)= -1.43074307E+01
================================================================================
== DATASET 3 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 3, }
dimensions: {natom: 4, nkpt: 10, mband: 26, nsppol: 2, nspinor: 1, nspden: 2, mpw: 376, }
cutoff_energies: {ecut: 10.0, pawecutdg: 20.0, }
electrons: {nelect: 4.80000000E+01, charge: 0.00000000E+00, occopt: 0.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 0, ionmov: 0, optcell: 0, iscf: 17, paral_kgb: 0, }
...
mkfilename: getwfk from: ./tlruj_1.o_WFK
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 3.9400000 3.9400000 7.8800000 G(1)= -0.0634518 -0.0634518 0.1903553
R(2)= 7.8800000 3.9400000 3.9400000 G(2)= 0.1903553 -0.0634518 -0.0634518
R(3)= 3.9400000 7.8800000 3.9400000 G(3)= -0.0634518 0.1903553 -0.0634518
Unit cell volume ucvol= 2.4465194E+02 bohr^3
Angles (23,13,12)= 3.35573098E+01 3.35573098E+01 3.35573098E+01 degrees
Coarse grid specifications (used for wave-functions):
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 30 30 30
ecut(hartree)= 10.000 => boxcut(ratio)= 2.18911
Fine grid specifications (used for densities):
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 40 40 40
ecut(hartree)= 20.000 => boxcut(ratio)= 2.06006
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file ./tlruj_1.o_WFK
_setup2: Arith. and geom. avg. npw (full set) are 368.875 368.830
******************************************
DFT+U Method used: FLL
******************************************
================================================================================
--- !BeginCycle
iteration_state: {dtset: 3, }
solver: {iscf: 17, nstep: 30, nline: 4, wfoptalg: 10, }
tolerances: {tolvrs: 1.00E-09, }
...
iter Etot(hartree) deltaE(h) residm nres2 difmagsph maxmagsph
ETOT 1 -363.72671779665 -3.637E+02 1.318E-08 4.640E-02 1.555E-01 1.555E-01
ETOT 2 -363.72727647899 -5.587E-04 1.276E-07 1.331E+00 4.058E-02 1.166E-01
ETOT 3 -363.72668375692 5.927E-04 4.331E-05 4.581E-04 4.078E-02 1.557E-01
ETOT 4 -363.72668525273 -1.496E-06 8.870E-08 3.588E-05 1.214E-04 1.558E-01
ETOT 5 -363.72668532365 -7.092E-08 1.599E-08 1.232E-04 1.129E-04 1.559E-01
ETOT 6 -363.72668529027 3.338E-08 1.532E-09 3.746E-05 6.651E-05 1.560E-01
ETOT 7 -363.72668528382 6.450E-09 1.557E-09 9.362E-06 6.721E-05 1.561E-01
ETOT 8 -363.72668528813 -4.312E-09 1.007E-10 6.943E-06 6.646E-05 1.561E-01
ETOT 9 -363.72668529147 -3.337E-09 4.036E-10 3.772E-06 1.090E-04 1.562E-01
ETOT 10 -363.72668529410 -2.625E-09 2.280E-10 1.805E-06 1.540E-04 1.564E-01
ETOT 11 -363.72668529467 -5.779E-10 8.387E-10 1.256E-07 2.173E-04 1.566E-01
ETOT 12 -363.72668529590 -1.222E-09 8.610E-11 2.855E-08 7.026E-05 1.567E-01
ETOT 13 -363.72668529637 -4.765E-10 1.252E-10 2.086E-08 1.949E-05 1.567E-01
ETOT 14 -363.72668529649 -1.164E-10 1.224E-11 1.654E-09 1.729E-05 1.567E-01
ETOT 15 -363.72668529651 -2.558E-11 1.273E-11 3.676E-10 6.833E-06 1.567E-01
At SCF step 15 nres2 = 3.68E-10 < tolvrs= 1.00E-09 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42475182E-01 sigma(3 2)= -4.87136612E-04
sigma(2 2)= 1.42475182E-01 sigma(3 1)= -4.87136612E-04
sigma(3 3)= 1.42475182E-01 sigma(2 1)= -4.87136612E-04
--- !ResultsGS
iteration_state: {dtset: 3, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.9400000, 3.9400000, 7.8800000, ]
- [ 7.8800000, 3.9400000, 3.9400000, ]
- [ 3.9400000, 7.8800000, 3.9400000, ]
lattice_lengths: [ 9.65099, 9.65099, 9.65099, ]
lattice_angles: [ 33.557, 33.557, 33.557, ] # degrees, (23, 13, 12)
lattice_volume: 2.4465194E+02
convergence: {deltae: -2.558E-11, res2: 3.676E-10, residm: 1.273E-11, diffor: null, }
etotal : -3.63726685E+02
entropy : 0.00000000E+00
fermie : 4.07678431E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.42475182E-01, -4.87136612E-04, -4.87136612E-04, ]
- [ -4.87136612E-04, 1.42475182E-01, -4.87136612E-04, ]
- [ -4.87136612E-04, -4.87136612E-04, 1.42475182E-01, ]
pressure_GPa: -4.1918E+03
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Ni]
- [ 5.0000E-01, 5.0000E-01, 5.0000E-01, Ni]
- [ 2.5000E-01, 2.5000E-01, 2.5000E-01, O]
- [ 7.5000E-01, 7.5000E-01, 7.5000E-01, O]
cartesian_forces: # hartree/bohr
- [ 6.77626358E-21, -2.03287907E-20, 6.77626358E-21, ]
- [ 6.77626358E-21, -2.03287907E-20, 6.77626358E-21, ]
- [ -1.30232979E-04, -1.30232979E-04, -1.30232979E-04, ]
- [ 1.30232979E-04, 1.30232979E-04, 1.30232979E-04, ]
force_length_stats: {min: 2.24743238E-20, max: 2.25570136E-04, mean: 1.12785068E-04, }
...
Integrated electronic and magnetization densities in atomic spheres:
------------------------------------------------------------------------------------------------------------
Radius=ratsph(iatom), smearing ratsm= 0.0000. Diff(up-dn)=approximate z local magnetic moment.
Atom Radius up_density dn_density Total(up+dn) Diff(up-dn)
1 Ni 1.81432 8.024974 7.868389 15.893363 0.156585
2 Ni 1.81432 7.866410 8.023142 15.889551 -0.156732
3 O 1.41465 2.292268 2.292194 4.584462 0.000074
4 O 1.41465 2.292268 2.292194 4.584462 0.000074
------------------------------------------------------------------------------------------------------------
Sum: 20.475920 20.475919 40.951839 0.000001
Total magnetization (from the atomic spheres): 0.000001
Total magnetization (exact up - dn): -0.000000
PAW TEST:
==== Compensation charge inside spheres ============
The following values must be close to each other ...
Compensation charge over spherical meshes = 12.719705992573743
Compensation charge over fine fft grid = 12.719993068861752
==== Results concerning PAW augmentation regions ====
Total pseudopotential strength Dij (hartree):
Atom # 1 - Spin component 1
-2.33054 0.37546 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00170 -0.00170 0.00000 -0.00170 ...
0.37546 0.04446 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00034 0.00034 0.00000 0.00034 ...
0.00000 0.00000 -2.24190 -0.00126 -0.00126 0.82140 0.00077 0.00077 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00126 -2.24190 -0.00126 0.00077 0.82140 0.00077 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00126 -0.00126 -2.24190 0.00077 0.00077 0.82140 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.82140 0.00077 0.00077 0.37067 -0.00046 -0.00046 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00077 0.82140 0.00077 -0.00046 0.37067 -0.00046 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00077 0.00077 0.82140 -0.00046 -0.00046 0.37067 0.00000 0.00000 0.00000 0.00000 ...
-0.00170 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.91232 -0.00101 0.00094 -0.00101 ...
-0.00170 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00101 -0.91232 -0.00047 -0.00101 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00094 -0.00047 -0.91360 -0.00047 ...
-0.00170 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00101 -0.00101 -0.00047 -0.91232 ...
... only 12 components have been written...
Atom # 1 - Spin component 2
-2.32675 0.37475 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00223 -0.00223 0.00000 -0.00223 ...
0.37475 0.04457 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00044 0.00044 0.00000 0.00044 ...
0.00000 0.00000 -2.23873 -0.00165 -0.00165 0.81962 0.00099 0.00099 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00165 -2.23873 -0.00165 0.00099 0.81962 0.00099 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00165 -0.00165 -2.23873 0.00099 0.00099 0.81962 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.81962 0.00099 0.00099 0.37173 -0.00060 -0.00060 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00099 0.81962 0.00099 -0.00060 0.37173 -0.00060 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00099 0.00099 0.81962 -0.00060 -0.00060 0.37173 0.00000 0.00000 0.00000 0.00000 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.90957 -0.00114 0.00137 -0.00114 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00114 -0.90957 -0.00068 -0.00114 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00137 -0.00068 -0.91066 -0.00068 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00114 -0.00114 -0.00068 -0.90957 ...
... only 12 components have been written...
Atom # 4 - Spin component 1
0.61340 -1.75562 0.00001 0.00001 0.00001 -0.00007 -0.00007 -0.00007
-1.75562 5.08527 -0.00003 -0.00003 -0.00003 0.00017 0.00017 0.00017
0.00001 -0.00003 -0.29402 -0.00002 -0.00002 0.88843 0.00011 0.00011
0.00001 -0.00003 -0.00002 -0.29402 -0.00002 0.00011 0.88843 0.00011
0.00001 -0.00003 -0.00002 -0.00002 -0.29402 0.00011 0.00011 0.88843
-0.00007 0.00017 0.88843 0.00011 0.00011 0.11845 -0.00056 -0.00056
-0.00007 0.00017 0.00011 0.88843 0.00011 -0.00056 0.11845 -0.00056
-0.00007 0.00017 0.00011 0.00011 0.88843 -0.00056 -0.00056 0.11845
Atom # 4 - Spin component 2
0.61340 -1.75561 -0.00001 -0.00001 -0.00001 0.00009 0.00009 0.00009
-1.75561 5.08525 0.00004 0.00004 0.00004 -0.00024 -0.00024 -0.00024
-0.00001 0.00004 -0.29402 -0.00002 -0.00002 0.88842 0.00011 0.00011
-0.00001 0.00004 -0.00002 -0.29402 -0.00002 0.00011 0.88842 0.00011
-0.00001 0.00004 -0.00002 -0.00002 -0.29402 0.00011 0.00011 0.88842
0.00009 -0.00024 0.88842 0.00011 0.00011 0.11849 -0.00056 -0.00056
0.00009 -0.00024 0.00011 0.88842 0.00011 -0.00056 0.11849 -0.00056
0.00009 -0.00024 0.00011 0.00011 0.88842 -0.00056 -0.00056 0.11849
Augmentation waves occupancies Rhoij:
Atom # 1 - Spin component 1
0.97694 -0.16681 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00014 -0.00014 0.00000 -0.00014 ...
-0.16681 0.45824 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00358 -0.00358 0.00000 -0.00358 ...
0.00000 0.00000 1.06704 0.00002 0.00002 0.13577 0.00008 0.00008 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00002 1.06704 0.00002 0.00008 0.13577 0.00008 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00002 0.00002 1.06704 0.00008 0.00008 0.13577 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.13577 0.00008 0.00008 0.02463 0.00002 0.00002 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00008 0.13577 0.00008 0.00002 0.02463 0.00002 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00008 0.00008 0.13577 0.00002 0.00002 0.02463 0.00000 0.00000 0.00000 0.00000 ...
-0.00014 -0.00358 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 1.15501 -0.02174 0.02664 -0.02174 ...
-0.00014 -0.00358 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.02174 1.15501 -0.01332 -0.02174 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.02664 -0.01332 1.05965 -0.01332 ...
-0.00014 -0.00358 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.02174 -0.02174 -0.01332 1.15501 ...
... only 12 components have been written...
Atom # 1 - Spin component 2
0.97687 -0.16733 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00013 -0.00013 0.00000 -0.00013 ...
-0.16733 0.45666 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00128 -0.00128 0.00000 -0.00128 ...
0.00000 0.00000 1.06701 0.00003 0.00003 0.13566 0.00012 0.00012 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00003 1.06701 0.00003 0.00012 0.13566 0.00012 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00003 0.00003 1.06701 0.00012 0.00012 0.13566 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.13566 0.00012 0.00012 0.02455 0.00004 0.00004 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00012 0.13566 0.00012 0.00004 0.02455 0.00004 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00012 0.00012 0.13566 0.00004 0.00004 0.02455 0.00000 0.00000 0.00000 0.00000 ...
-0.00013 -0.00128 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 1.11070 -0.01390 0.00067 -0.01390 ...
-0.00013 -0.00128 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.01390 1.11070 -0.00033 -0.01390 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00067 -0.00033 1.01216 -0.00033 ...
-0.00013 -0.00128 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.01390 -0.01390 -0.00033 1.11070 ...
... only 12 components have been written...
Atom # 4 - Spin component 1
1.01157 0.01395 -0.00077 -0.00077 -0.00077 0.00003 0.00003 0.00003
0.01395 0.00119 0.00013 0.00013 0.00013 -0.00000 -0.00000 -0.00000
-0.00077 0.00013 1.06827 -0.00321 -0.00321 0.05052 -0.00033 -0.00033
-0.00077 0.00013 -0.00321 1.06827 -0.00321 -0.00033 0.05052 -0.00033
-0.00077 0.00013 -0.00321 -0.00321 1.06827 -0.00033 -0.00033 0.05052
0.00003 -0.00000 0.05052 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
0.00003 -0.00000 -0.00033 0.05052 -0.00033 -0.00001 0.00287 -0.00001
0.00003 -0.00000 -0.00033 -0.00033 0.05052 -0.00001 -0.00001 0.00287
Atom # 4 - Spin component 2
1.01155 0.01396 0.00067 0.00067 0.00067 -0.00003 -0.00003 -0.00003
0.01396 0.00119 -0.00011 -0.00011 -0.00011 0.00000 0.00000 0.00000
0.00067 -0.00011 1.06824 -0.00320 -0.00320 0.05052 -0.00033 -0.00033
0.00067 -0.00011 -0.00320 1.06824 -0.00320 -0.00033 0.05052 -0.00033
0.00067 -0.00011 -0.00320 -0.00320 1.06824 -0.00033 -0.00033 0.05052
-0.00003 0.00000 0.05052 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00003 0.00000 -0.00033 0.05052 -0.00033 -0.00001 0.00287 -0.00001
-0.00003 0.00000 -0.00033 -0.00033 0.05052 -0.00001 -0.00001 0.00287
"PAW+U" part of augmentation waves occupancies Rhoij:
Atom # 1 - L=2 ONLY - Spin component 1
1.15501 -0.02174 0.02664 -0.02174 0.00000 0.42939 -0.00883 0.01033 -0.00883 0.00000
-0.02174 1.15501 -0.01332 -0.02174 0.02307 -0.00883 0.42939 -0.00516 -0.00883 0.00894
0.02664 -0.01332 1.05965 -0.01332 0.00000 0.01204 -0.00602 0.39188 -0.00602 0.00000
-0.02174 -0.02174 -0.01332 1.15501 -0.02307 -0.00883 -0.00883 -0.00516 0.42939 -0.00894
0.00000 0.02307 0.00000 -0.02307 1.05965 0.00000 0.01042 0.00000 -0.01042 0.39188
0.42939 -0.00883 0.01204 -0.00883 0.00000 0.16420 -0.00362 0.00472 -0.00362 0.00000
-0.00883 0.42939 -0.00602 -0.00883 0.01042 -0.00362 0.16420 -0.00236 -0.00362 0.00409
0.01033 -0.00516 0.39188 -0.00516 0.00000 0.00472 -0.00236 0.15191 -0.00236 0.00000
-0.00883 -0.00883 -0.00602 0.42939 -0.01042 -0.00362 -0.00362 -0.00236 0.16420 -0.00409
0.00000 0.00894 0.00000 -0.00894 0.39188 0.00000 0.00409 0.00000 -0.00409 0.15191
Atom # 1 - L=2 ONLY - Spin component 2
1.11070 -0.01390 0.00067 -0.01390 0.00000 0.40914 -0.00545 -0.00019 -0.00545 0.00000
-0.01390 1.11070 -0.00033 -0.01390 0.00058 -0.00545 0.40914 0.00010 -0.00545 -0.00017
0.00067 -0.00033 1.01216 -0.00033 0.00000 -0.00039 0.00020 0.37384 0.00020 0.00000
-0.01390 -0.01390 -0.00033 1.11070 -0.00058 -0.00545 -0.00545 0.00010 0.40914 0.00017
0.00000 0.00058 0.00000 -0.00058 1.01216 0.00000 -0.00034 0.00000 0.00034 0.37384
0.40914 -0.00545 -0.00039 -0.00545 0.00000 0.15494 -0.00217 -0.00030 -0.00217 0.00000
-0.00545 0.40914 0.00020 -0.00545 -0.00034 -0.00217 0.15494 0.00015 -0.00217 -0.00026
-0.00019 0.00010 0.37384 0.00010 0.00000 -0.00030 0.00015 0.14502 0.00015 0.00000
-0.00545 -0.00545 0.00020 0.40914 0.00034 -0.00217 -0.00217 0.00015 0.15494 0.00026
0.00000 -0.00017 0.00000 0.00017 0.37384 0.00000 -0.00026 0.00000 0.00026 0.14502
Atom # 2 - L=2 ONLY - Spin component 1
1.11008 -0.01391 0.00060 -0.01391 0.00000 0.40882 -0.00546 -0.00023 -0.00546 0.00000
-0.01391 1.11008 -0.00030 -0.01391 0.00052 -0.00546 0.40882 0.00012 -0.00546 -0.00020
0.00060 -0.00030 1.01125 -0.00030 0.00000 -0.00045 0.00022 0.37346 0.00022 0.00000
-0.01391 -0.01391 -0.00030 1.11008 -0.00052 -0.00546 -0.00546 0.00012 0.40882 0.00020
0.00000 0.00052 0.00000 -0.00052 1.01125 0.00000 -0.00039 0.00000 0.00039 0.37346
0.40882 -0.00546 -0.00045 -0.00546 0.00000 0.15479 -0.00217 -0.00032 -0.00217 0.00000
-0.00546 0.40882 0.00022 -0.00546 -0.00039 -0.00217 0.15479 0.00016 -0.00217 -0.00028
-0.00023 0.00012 0.37346 0.00012 0.00000 -0.00032 0.00016 0.14487 0.00016 0.00000
-0.00546 -0.00546 0.00022 0.40882 0.00039 -0.00217 -0.00217 0.00016 0.15479 0.00028
0.00000 -0.00020 0.00000 0.00020 0.37346 0.00000 -0.00028 0.00000 0.00028 0.14487
Atom # 2 - L=2 ONLY - Spin component 2
1.15450 -0.02174 0.02658 -0.02174 0.00000 0.42913 -0.00883 0.01029 -0.00883 0.00000
-0.02174 1.15450 -0.01329 -0.02174 0.02302 -0.00883 0.42913 -0.00514 -0.00883 0.00891
0.02658 -0.01329 1.05871 -0.01329 0.00000 0.01199 -0.00600 0.39148 -0.00600 0.00000
-0.02174 -0.02174 -0.01329 1.15450 -0.02302 -0.00883 -0.00883 -0.00514 0.42913 -0.00891
0.00000 0.02302 0.00000 -0.02302 1.05871 0.00000 0.01038 0.00000 -0.01038 0.39148
0.42913 -0.00883 0.01199 -0.00883 0.00000 0.16409 -0.00362 0.00470 -0.00362 0.00000
-0.00883 0.42913 -0.00600 -0.00883 0.01038 -0.00362 0.16409 -0.00235 -0.00362 0.00407
0.01029 -0.00514 0.39148 -0.00514 0.00000 0.00470 -0.00235 0.15176 -0.00235 0.00000
-0.00883 -0.00883 -0.00600 0.42913 -0.01038 -0.00362 -0.00362 -0.00235 0.16409 -0.00407
0.00000 0.00891 0.00000 -0.00891 0.39148 0.00000 0.00407 0.00000 -0.00407 0.15176
Atom # 3 - L=1 ONLY - Spin component 1
1.06827 -0.00321 -0.00321 0.05052 -0.00033 -0.00033
-0.00321 1.06827 -0.00321 -0.00033 0.05052 -0.00033
-0.00321 -0.00321 1.06827 -0.00033 -0.00033 0.05052
0.05052 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05052 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05052 -0.00001 -0.00001 0.00287
Atom # 3 - L=1 ONLY - Spin component 2
1.06824 -0.00320 -0.00320 0.05052 -0.00033 -0.00033
-0.00320 1.06824 -0.00320 -0.00033 0.05052 -0.00033
-0.00320 -0.00320 1.06824 -0.00033 -0.00033 0.05052
0.05052 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05052 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05052 -0.00001 -0.00001 0.00287
Atom # 4 - L=1 ONLY - Spin component 1
1.06827 -0.00321 -0.00321 0.05052 -0.00033 -0.00033
-0.00321 1.06827 -0.00321 -0.00033 0.05052 -0.00033
-0.00321 -0.00321 1.06827 -0.00033 -0.00033 0.05052
0.05052 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05052 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05052 -0.00001 -0.00001 0.00287
Atom # 4 - L=1 ONLY - Spin component 2
1.06824 -0.00320 -0.00320 0.05052 -0.00033 -0.00033
-0.00320 1.06824 -0.00320 -0.00033 0.05052 -0.00033
-0.00320 -0.00320 1.06824 -0.00033 -0.00033 0.05052
0.05052 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05052 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05052 -0.00001 -0.00001 0.00287
---------- DFT+U DATA ---------------------------------------------------
====== For Atom 1, occupations for correlated orbitals. lpawu = 2
Atom 1. Occ. for lpawu and for spin 1 = 4.40952
Atom 1. Occ. for lpawu and for spin 2 = 4.23323
=> On atom 1, local Mag. for lpawu is -0.176283
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.91173 -0.01694 0.02064 -0.01694 0.00000
-0.01694 0.91173 -0.01032 -0.01694 0.01788
0.02064 -0.01032 0.83716 -0.01032 0.00000
-0.01694 -0.01694 -0.01032 0.91173 -0.01788
0.00000 0.01788 0.00000 -0.01788 0.83716
Occupation matrix for spin 2
0.87789 -0.01089 0.00069 -0.01089 0.00000
-0.01089 0.87789 -0.00034 -0.01089 0.00059
0.00069 -0.00034 0.79979 -0.00034 0.00000
-0.01089 -0.01089 -0.00034 0.87789 -0.00059
0.00000 0.00059 0.00000 -0.00059 0.79979
====== For Atom 2, occupations for correlated orbitals. lpawu = 2
Atom 2. Occ. for lpawu and for spin 1 = 4.23043
Atom 2. Occ. for lpawu and for spin 2 = 4.40690
=> On atom 2, local Mag. for lpawu is 0.176472
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.87742 -0.01089 0.00065 -0.01089 0.00000
-0.01089 0.87742 -0.00032 -0.01089 0.00056
0.00065 -0.00032 0.79909 -0.00032 0.00000
-0.01089 -0.01089 -0.00032 0.87742 -0.00056
0.00000 0.00056 0.00000 -0.00056 0.79909
Occupation matrix for spin 2
0.91135 -0.01694 0.02060 -0.01694 0.00000
-0.01694 0.91135 -0.01030 -0.01694 0.01784
0.02060 -0.01030 0.83643 -0.01030 0.00000
-0.01694 -0.01694 -0.01030 0.91135 -0.01784
0.00000 0.01784 0.00000 -0.01784 0.83643
====== For Atom 3, occupations for correlated orbitals. lpawu = 1
Atom 3. Occ. for lpawu and for spin 1 = 1.55465
Atom 3. Occ. for lpawu and for spin 2 = 1.55461
=> On atom 3, local Mag. for lpawu is -0.000048
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.51822 -0.00082 -0.00082
-0.00082 0.51822 -0.00082
-0.00082 -0.00082 0.51822
Occupation matrix for spin 2
0.51820 -0.00081 -0.00081
-0.00081 0.51820 -0.00081
-0.00081 -0.00081 0.51820
====== For Atom 4, occupations for correlated orbitals. lpawu = 1
Atom 4. Occ. for lpawu and for spin 1 = 1.55465
Atom 4. Occ. for lpawu and for spin 2 = 1.55461
=> On atom 4, local Mag. for lpawu is -0.000048
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.51822 -0.00082 -0.00082
-0.00082 0.51822 -0.00082
-0.00082 -0.00082 0.51822
Occupation matrix for spin 2
0.51820 -0.00081 -0.00081
-0.00081 0.51820 -0.00081
-0.00081 -0.00081 0.51820
---------- calculate U, (J) start ----------
All pawujat ok and equal to 1
macro_uj=1 and nspden=2: standard determination of Hubbard U-parameter
*********************************************************************
************************ Linear Response U ************************
Info printed for perturbed atom: 1
Perturbations Occupations
--------------- -----------------------------
alpha [eV] Unscreened Screened
--------------- -----------------------------
0.0000000000 8.6380182458 8.6380182458
-0.0500000000 8.6560314451 8.6427490198
Scalar response functions:
Chi0 [eV^-1]: -0.80059
Chi [eV^-1]: -0.09462
The scalar U from the two-point regression scheme is 9.32001 eV.
*********************************************************************
*********************************************************************
Note: For more reliable linear regressions of the response
matrices, it is advised that you have more than two points.
See the LRUJ protocol for more information.
URES ii nat r_max U(J)[eV] U_ASA[eV] U_inf[eV]
URES 1 1 0.00000 2.33000 1.87503 1.81378
URES 2 8 11.14400 8.16939 6.57418 6.35943
URES 3 27 12.45940 8.97527 7.22270 6.98675
URES 4 64 22.28800 9.17442 7.38296 7.14179
URES 5 125 24.28780 9.24546 7.44013 7.19708
URES 6 216 33.43200 9.27686 7.46540 7.22153
---------- calculate U, (J) end --------------
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 10.176E-14; max= 12.732E-12
reduced coordinates (array xred) for 4 atoms
0.000000000000 0.000000000000 0.000000000000
0.500000000000 0.500000000000 0.500000000000
0.250000000000 0.250000000000 0.250000000000
0.750000000000 0.750000000000 0.750000000000
rms dE/dt= 1.4513E-03; max dE/dt= 2.0525E-03; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
2 0.000000000000 0.000000000000 0.000000000000
3 0.002052471748 0.002052471748 0.002052471748
4 -0.002052471748 -0.002052471748 -0.002052471748
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
2 4.16991641908672 4.16991641908672 4.16991641908672
3 2.08495820954336 2.08495820954336 2.08495820954336
4 6.25487462863008 6.25487462863008 6.25487462863008
cartesian forces (hartree/bohr) at end:
1 0.00000000000000 -0.00000000000000 0.00000000000000
2 0.00000000000000 -0.00000000000000 0.00000000000000
3 -0.00013023297894 -0.00013023297894 -0.00013023297894
4 0.00013023297894 0.00013023297894 0.00013023297894
frms,max,avg= 9.2088623E-05 1.3023298E-04 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 0.00000000000000 -0.00000000000000 0.00000000000000
2 0.00000000000000 -0.00000000000000 0.00000000000000
3 -0.00669684903543 -0.00669684903543 -0.00669684903543
4 0.00669684903543 0.00669684903543 0.00669684903543
frms,max,avg= 4.7353874E-03 6.6968490E-03 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.880000000000 7.880000000000 7.880000000000 bohr
= 4.169916419087 4.169916419087 4.169916419087 angstroms
Fermi (or HOMO) energy (hartree) = 0.40768 Average Vxc (hartree)= -0.46905
Eigenvalues (hartree) for nkpt= 10 k points, SPIN UP:
kpt# 1, nband= 26, wtk= 0.03125, kpt= 0.1250 0.1250 0.1250 (reduced coord)
-4.13990 -4.13702 -2.37077 -2.33851 -2.33184 -2.33184 -2.30940 -2.30940
-0.40334 -0.35615 0.07022 0.21096 0.21096 0.22655 0.22655 0.25279
0.26163 0.26163 0.31910 0.31910 0.37475 0.38738 0.38738 0.39683
0.40071 0.43654
prteigrs : prtvol=0 or 1, do not print more k-points.
Eigenvalues (hartree) for nkpt= 10 k points, SPIN DOWN:
kpt# 1, nband= 26, wtk= 0.03125, kpt= 0.1250 0.1250 0.1250 (reduced coord)
-4.14228 -4.13465 -2.37103 -2.33825 -2.33246 -2.33246 -2.30878 -2.30878
-0.40333 -0.35614 0.07022 0.21097 0.21097 0.22655 0.22655 0.25279
0.26163 0.26163 0.31910 0.31910 0.37478 0.38739 0.38739 0.39683
0.40069 0.43653
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 3, }
comment : Components of total free energy in Hartree
kinetic : 8.94818960902317E+01
hartree : 8.65592597737557E+01
xc : -2.18491558913554E+01
Ewald energy : -2.39331554544165E+02
psp_core : 1.04313020493122E+01
local_psp : -2.68603774708667E+02
PAW spherical_terms : -2.03987767340905E+01
total_energy : -3.63710803964979E+02
total_energy_eV : -9.89707516852731E+03
...
--- !EnergyTermsDC
iteration_state : {dtset: 3, }
comment : '"Double-counting" decomposition of free energy'
band_energy : -4.00290379975472E+01
Ewald energy : -2.39331554544165E+02
psp_core : 1.04313020493122E+01
xc_dc : -7.99992731682520E+01
PAW spherical_terms : -1.47981216358621E+01
total_energy_dc : -3.63726685296514E+02
total_energy_dc_eV : -9.89750732157383E+03
...
--- !EnergyTermsWithPAWCore
iteration_state : {dtset: 3, }
comment : 'Components of total free energy, including PAW core contributions'
Total energy : -3.18179009640906E+03
Total energy DC : -3.18180597774060E+03
Total energy (eV) : -8.65809192670241E+04
Total energy DC (eV): -8.65813514200706E+04
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42475182E-01 sigma(3 2)= -4.87136612E-04
sigma(2 2)= 1.42475182E-01 sigma(3 1)= -4.87136612E-04
sigma(3 3)= 1.42475182E-01 sigma(2 1)= -4.87136612E-04
-Cartesian components of stress tensor (GPa) [Pressure= -4.1918E+03 GPa]
- sigma(1 1)= 4.19176458E+03 sigma(3 2)= -1.43320539E+01
- sigma(2 2)= 4.19176458E+03 sigma(3 1)= -1.43320539E+01
- sigma(3 3)= 4.19176458E+03 sigma(2 1)= -1.43320539E+01
================================================================================
== DATASET 4 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 4, }
dimensions: {natom: 4, nkpt: 10, mband: 26, nsppol: 2, nspinor: 1, nspden: 2, mpw: 376, }
cutoff_energies: {ecut: 10.0, pawecutdg: 20.0, }
electrons: {nelect: 4.80000000E+01, charge: 0.00000000E+00, occopt: 0.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 0, ionmov: 0, optcell: 0, iscf: 17, paral_kgb: 0, }
...
mkfilename: getwfk from: ./tlruj_1.o_WFK
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 3.9400000 3.9400000 7.8800000 G(1)= -0.0634518 -0.0634518 0.1903553
R(2)= 7.8800000 3.9400000 3.9400000 G(2)= 0.1903553 -0.0634518 -0.0634518
R(3)= 3.9400000 7.8800000 3.9400000 G(3)= -0.0634518 0.1903553 -0.0634518
Unit cell volume ucvol= 2.4465194E+02 bohr^3
Angles (23,13,12)= 3.35573098E+01 3.35573098E+01 3.35573098E+01 degrees
Coarse grid specifications (used for wave-functions):
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 30 30 30
ecut(hartree)= 10.000 => boxcut(ratio)= 2.18911
Fine grid specifications (used for densities):
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 40 40 40
ecut(hartree)= 20.000 => boxcut(ratio)= 2.06006
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file ./tlruj_1.o_WFK
_setup2: Arith. and geom. avg. npw (full set) are 368.875 368.830
******************************************
DFT+U Method used: FLL
******************************************
================================================================================
--- !BeginCycle
iteration_state: {dtset: 4, }
solver: {iscf: 17, nstep: 30, nline: 4, wfoptalg: 10, }
tolerances: {tolvrs: 1.00E-09, }
...
iter Etot(hartree) deltaE(h) residm nres2 difmagsph maxmagsph
ETOT 1 -363.69497467175 -3.637E+02 1.422E-08 5.078E-02 1.564E-01 1.564E-01
ETOT 2 -363.69557245658 -5.978E-04 1.316E-07 1.168E+00 4.392E-02 1.154E-01
ETOT 3 -363.69494016024 6.323E-04 4.555E-05 1.072E-03 4.345E-02 1.560E-01
ETOT 4 -363.69494111179 -9.515E-07 9.840E-08 3.917E-05 3.746E-04 1.563E-01
ETOT 5 -363.69494117386 -6.208E-08 3.073E-08 6.116E-05 1.002E-04 1.564E-01
ETOT 6 -363.69494118860 -1.473E-08 1.708E-09 3.891E-05 1.983E-04 1.566E-01
ETOT 7 -363.69494117792 1.067E-08 2.340E-09 7.587E-06 1.577E-04 1.568E-01
ETOT 8 -363.69494117774 1.797E-10 3.733E-11 7.714E-06 5.511E-06 1.568E-01
ETOT 9 -363.69494118171 -3.970E-09 9.901E-11 8.341E-06 5.028E-05 1.568E-01
ETOT 10 -363.69494119220 -1.049E-08 9.398E-10 3.229E-06 3.026E-04 1.571E-01
ETOT 11 -363.69494119994 -7.737E-09 1.473E-09 7.507E-07 5.147E-04 1.576E-01
ETOT 12 -363.69494120187 -1.936E-09 1.150E-10 1.973E-07 1.125E-04 1.577E-01
ETOT 13 -363.69494120467 -2.798E-09 4.707E-10 1.221E-07 1.358E-04 1.579E-01
ETOT 14 -363.69494120503 -3.591E-10 3.785E-11 9.363E-08 1.001E-05 1.579E-01
ETOT 15 -363.69494120531 -2.793E-10 2.791E-11 1.451E-08 1.111E-05 1.579E-01
ETOT 16 -363.69494120543 -1.166E-10 1.069E-11 9.931E-09 1.306E-05 1.579E-01
ETOT 17 -363.69494120548 -5.144E-11 5.382E-12 3.810E-10 9.806E-06 1.579E-01
At SCF step 17 nres2 = 3.81E-10 < tolvrs= 1.00E-09 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42464714E-01 sigma(3 2)= -4.89478733E-04
sigma(2 2)= 1.42464714E-01 sigma(3 1)= -4.89478733E-04
sigma(3 3)= 1.42464714E-01 sigma(2 1)= -4.89478733E-04
--- !ResultsGS
iteration_state: {dtset: 4, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.9400000, 3.9400000, 7.8800000, ]
- [ 7.8800000, 3.9400000, 3.9400000, ]
- [ 3.9400000, 7.8800000, 3.9400000, ]
lattice_lengths: [ 9.65099, 9.65099, 9.65099, ]
lattice_angles: [ 33.557, 33.557, 33.557, ] # degrees, (23, 13, 12)
lattice_volume: 2.4465194E+02
convergence: {deltae: -5.144E-11, res2: 3.810E-10, residm: 5.382E-12, diffor: null, }
etotal : -3.63694941E+02
entropy : 0.00000000E+00
fermie : 4.08504527E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.42464714E-01, -4.89478733E-04, -4.89478733E-04, ]
- [ -4.89478733E-04, 1.42464714E-01, -4.89478733E-04, ]
- [ -4.89478733E-04, -4.89478733E-04, 1.42464714E-01, ]
pressure_GPa: -4.1915E+03
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Ni]
- [ 5.0000E-01, 5.0000E-01, 5.0000E-01, Ni]
- [ 2.5000E-01, 2.5000E-01, 2.5000E-01, O]
- [ 7.5000E-01, 7.5000E-01, 7.5000E-01, O]
cartesian_forces: # hartree/bohr
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ -0.00000000E+00, -0.00000000E+00, -0.00000000E+00, ]
- [ 1.32510313E-04, 1.32510313E-04, 1.32510313E-04, ]
- [ -1.32510313E-04, -1.32510313E-04, -1.32510313E-04, ]
force_length_stats: {min: 0.00000000E+00, max: 2.29514595E-04, mean: 1.14757297E-04, }
...
Integrated electronic and magnetization densities in atomic spheres:
------------------------------------------------------------------------------------------------------------
Radius=ratsph(iatom), smearing ratsm= 0.0000. Diff(up-dn)=approximate z local magnetic moment.
Atom Radius up_density dn_density Total(up+dn) Diff(up-dn)
1 Ni 1.81432 8.022336 7.864412 15.886747 0.157924
2 Ni 1.81432 7.866440 8.024121 15.890561 -0.157681
3 O 1.41465 2.292330 2.292422 4.584752 -0.000092
4 O 1.41465 2.292330 2.292422 4.584752 -0.000092
------------------------------------------------------------------------------------------------------------
Sum: 20.473436 20.473377 40.946813 0.000059
Total magnetization (from the atomic spheres): 0.000059
Total magnetization (exact up - dn): 0.000000
PAW TEST:
==== Compensation charge inside spheres ============
The following values must be close to each other ...
Compensation charge over spherical meshes = 12.717893954276752
Compensation charge over fine fft grid = 12.718180257672545
==== Results concerning PAW augmentation regions ====
Total pseudopotential strength Dij (hartree):
Atom # 1 - Spin component 1
-2.33247 0.37596 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00171 -0.00171 0.00000 -0.00171 ...
0.37596 0.04437 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00034 0.00034 0.00000 0.00034 ...
0.00000 0.00000 -2.24365 -0.00127 -0.00127 0.82275 0.00077 0.00077 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00127 -2.24365 -0.00127 0.00077 0.82275 0.00077 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00127 -0.00127 -2.24365 0.00077 0.00077 0.82275 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.82275 0.00077 0.00077 0.36979 -0.00047 -0.00047 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00077 0.82275 0.00077 -0.00047 0.36979 -0.00047 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00077 0.00077 0.82275 -0.00047 -0.00047 0.36979 0.00000 0.00000 0.00000 0.00000 ...
-0.00171 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.91101 -0.00102 0.00094 -0.00102 ...
-0.00171 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00102 -0.91101 -0.00047 -0.00102 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00094 -0.00047 -0.91229 -0.00047 ...
-0.00171 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00102 -0.00102 -0.00047 -0.91101 ...
... only 12 components have been written...
Atom # 1 - Spin component 2
-2.32864 0.37525 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00223 -0.00223 0.00000 -0.00223 ...
0.37525 0.04448 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00044 0.00044 0.00000 0.00044 ...
0.00000 0.00000 -2.24045 -0.00165 -0.00165 0.82095 0.00099 0.00099 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00165 -2.24045 -0.00165 0.00099 0.82095 0.00099 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00165 -0.00165 -2.24045 0.00099 0.00099 0.82095 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.82095 0.00099 0.00099 0.37086 -0.00060 -0.00060 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00099 0.82095 0.00099 -0.00060 0.37086 -0.00060 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00099 0.00099 0.82095 -0.00060 -0.00060 0.37086 0.00000 0.00000 0.00000 0.00000 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.90823 -0.00114 0.00137 -0.00114 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00114 -0.90823 -0.00069 -0.00114 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00137 -0.00069 -0.90933 -0.00069 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00114 -0.00114 -0.00069 -0.90823 ...
... only 12 components have been written...
Atom # 4 - Spin component 1
0.61336 -1.75550 0.00002 0.00002 0.00002 -0.00010 -0.00010 -0.00010
-1.75550 5.08489 -0.00004 -0.00004 -0.00004 0.00025 0.00025 0.00025
0.00002 -0.00004 -0.29395 -0.00002 -0.00002 0.88808 0.00011 0.00011
0.00002 -0.00004 -0.00002 -0.29395 -0.00002 0.00011 0.88808 0.00011
0.00002 -0.00004 -0.00002 -0.00002 -0.29395 0.00011 0.00011 0.88808
-0.00010 0.00025 0.88808 0.00011 0.00011 0.12028 -0.00056 -0.00056
-0.00010 0.00025 0.00011 0.88808 0.00011 -0.00056 0.12028 -0.00056
-0.00010 0.00025 0.00011 0.00011 0.88808 -0.00056 -0.00056 0.12028
Atom # 4 - Spin component 2
0.61337 -1.75551 -0.00001 -0.00001 -0.00001 0.00007 0.00007 0.00007
-1.75551 5.08492 0.00003 0.00003 0.00003 -0.00019 -0.00019 -0.00019
-0.00001 0.00003 -0.29395 -0.00002 -0.00002 0.88809 0.00011 0.00011
-0.00001 0.00003 -0.00002 -0.29395 -0.00002 0.00011 0.88809 0.00011
-0.00001 0.00003 -0.00002 -0.00002 -0.29395 0.00011 0.00011 0.88809
0.00007 -0.00019 0.88809 0.00011 0.00011 0.12023 -0.00055 -0.00055
0.00007 -0.00019 0.00011 0.88809 0.00011 -0.00055 0.12023 -0.00055
0.00007 -0.00019 0.00011 0.00011 0.88809 -0.00055 -0.00055 0.12023
Augmentation waves occupancies Rhoij:
Atom # 1 - Spin component 1
0.97690 -0.16706 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00016 -0.00016 0.00000 -0.00016 ...
-0.16706 0.46117 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00409 -0.00409 0.00000 -0.00409 ...
0.00000 0.00000 1.06706 0.00002 0.00002 0.13566 0.00008 0.00008 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00002 1.06706 0.00002 0.00008 0.13566 0.00008 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00002 0.00002 1.06706 0.00008 0.00008 0.13566 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.13566 0.00008 0.00008 0.02463 0.00002 0.00002 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00008 0.13566 0.00008 0.00002 0.02463 0.00002 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00008 0.00008 0.13566 0.00002 0.00002 0.02463 0.00000 0.00000 0.00000 0.00000 ...
-0.00016 -0.00409 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 1.15418 -0.02175 0.02666 -0.02175 ...
-0.00016 -0.00409 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.02175 1.15418 -0.01333 -0.02175 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.02666 -0.01333 1.05835 -0.01333 ...
-0.00016 -0.00409 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.02175 -0.02175 -0.01333 1.15418 ...
... only 12 components have been written...
Atom # 1 - Spin component 2
0.97682 -0.16754 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00011 -0.00011 0.00000 -0.00011 ...
-0.16754 0.46102 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00055 -0.00055 0.00000 -0.00055 ...
0.00000 0.00000 1.06703 0.00003 0.00003 0.13555 0.00012 0.00012 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00003 1.06703 0.00003 0.00012 0.13555 0.00012 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00003 0.00003 1.06703 0.00012 0.00012 0.13555 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.13555 0.00012 0.00012 0.02455 0.00004 0.00004 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00012 0.13555 0.00012 0.00004 0.02455 0.00004 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00012 0.00012 0.13555 0.00004 0.00004 0.02455 0.00000 0.00000 0.00000 0.00000 ...
-0.00011 -0.00055 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 1.10934 -0.01422 0.00057 -0.01422 ...
-0.00011 -0.00055 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.01422 1.10934 -0.00029 -0.01422 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00057 -0.00029 1.01054 -0.00029 ...
-0.00011 -0.00055 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.01422 -0.01422 -0.00029 1.10934 ...
... only 12 components have been written...
Atom # 4 - Spin component 1
1.01160 0.01395 -0.00073 -0.00073 -0.00073 0.00003 0.00003 0.00003
0.01395 0.00120 0.00012 0.00012 0.00012 -0.00000 -0.00000 -0.00000
-0.00073 0.00012 1.06834 -0.00318 -0.00318 0.05053 -0.00033 -0.00033
-0.00073 0.00012 -0.00318 1.06834 -0.00318 -0.00033 0.05053 -0.00033
-0.00073 0.00012 -0.00318 -0.00318 1.06834 -0.00033 -0.00033 0.05053
0.00003 -0.00000 0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
0.00003 -0.00000 -0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
0.00003 -0.00000 -0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
Atom # 4 - Spin component 2
1.01162 0.01395 0.00083 0.00083 0.00083 -0.00003 -0.00003 -0.00003
0.01395 0.00120 -0.00015 -0.00015 -0.00015 -0.00000 -0.00000 -0.00000
0.00083 -0.00015 1.06837 -0.00319 -0.00319 0.05053 -0.00033 -0.00033
0.00083 -0.00015 -0.00319 1.06837 -0.00319 -0.00033 0.05053 -0.00033
0.00083 -0.00015 -0.00319 -0.00319 1.06837 -0.00033 -0.00033 0.05053
-0.00003 -0.00000 0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00003 -0.00000 -0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
-0.00003 -0.00000 -0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
"PAW+U" part of augmentation waves occupancies Rhoij:
Atom # 1 - L=2 ONLY - Spin component 1
1.15418 -0.02175 0.02666 -0.02175 0.00000 0.42901 -0.00884 0.01032 -0.00884 0.00000
-0.02175 1.15418 -0.01333 -0.02175 0.02309 -0.00884 0.42901 -0.00516 -0.00884 0.00894
0.02666 -0.01333 1.05835 -0.01333 0.00000 0.01204 -0.00602 0.39131 -0.00602 0.00000
-0.02175 -0.02175 -0.01333 1.15418 -0.02309 -0.00884 -0.00884 -0.00516 0.42901 -0.00894
0.00000 0.02309 0.00000 -0.02309 1.05835 0.00000 0.01042 0.00000 -0.01042 0.39131
0.42901 -0.00884 0.01204 -0.00884 0.00000 0.16405 -0.00362 0.00472 -0.00362 0.00000
-0.00884 0.42901 -0.00602 -0.00884 0.01042 -0.00362 0.16405 -0.00236 -0.00362 0.00408
0.01032 -0.00516 0.39131 -0.00516 0.00000 0.00472 -0.00236 0.15168 -0.00236 0.00000
-0.00884 -0.00884 -0.00602 0.42901 -0.01042 -0.00362 -0.00362 -0.00236 0.16405 -0.00408
0.00000 0.00894 0.00000 -0.00894 0.39131 0.00000 0.00408 0.00000 -0.00408 0.15168
Atom # 1 - L=2 ONLY - Spin component 2
1.10934 -0.01422 0.00057 -0.01422 0.00000 0.40850 -0.00560 -0.00025 -0.00560 0.00000
-0.01422 1.10934 -0.00029 -0.01422 0.00050 -0.00560 0.40850 0.00013 -0.00560 -0.00022
0.00057 -0.00029 1.01054 -0.00029 0.00000 -0.00046 0.00023 0.37315 0.00023 0.00000
-0.01422 -0.01422 -0.00029 1.10934 -0.00050 -0.00560 -0.00560 0.00013 0.40850 0.00022
0.00000 0.00050 0.00000 -0.00050 1.01054 0.00000 -0.00040 0.00000 0.00040 0.37315
0.40850 -0.00560 -0.00046 -0.00560 0.00000 0.15465 -0.00223 -0.00033 -0.00223 0.00000
-0.00560 0.40850 0.00023 -0.00560 -0.00040 -0.00223 0.15465 0.00017 -0.00223 -0.00029
-0.00025 0.00013 0.37315 0.00013 0.00000 -0.00033 0.00017 0.14474 0.00017 0.00000
-0.00560 -0.00560 0.00023 0.40850 0.00040 -0.00223 -0.00223 0.00017 0.15465 0.00029
0.00000 -0.00022 0.00000 0.00022 0.37315 0.00000 -0.00029 0.00000 0.00029 0.14474
Atom # 2 - L=2 ONLY - Spin component 1
1.11000 -0.01416 0.00063 -0.01416 0.00000 0.40884 -0.00557 -0.00021 -0.00557 0.00000
-0.01416 1.11000 -0.00032 -0.01416 0.00055 -0.00557 0.40884 0.00011 -0.00557 -0.00019
0.00063 -0.00032 1.01144 -0.00032 0.00000 -0.00041 0.00021 0.37353 0.00021 0.00000
-0.01416 -0.01416 -0.00032 1.11000 -0.00055 -0.00557 -0.00557 0.00011 0.40884 0.00019
0.00000 0.00055 0.00000 -0.00055 1.01144 0.00000 -0.00036 0.00000 0.00036 0.37353
0.40884 -0.00557 -0.00041 -0.00557 0.00000 0.15481 -0.00222 -0.00031 -0.00222 0.00000
-0.00557 0.40884 0.00021 -0.00557 -0.00036 -0.00222 0.15481 0.00015 -0.00222 -0.00027
-0.00021 0.00011 0.37353 0.00011 0.00000 -0.00031 0.00015 0.14489 0.00015 0.00000
-0.00557 -0.00557 0.00021 0.40884 0.00036 -0.00222 -0.00222 0.00015 0.15481 0.00027
0.00000 -0.00019 0.00000 0.00019 0.37353 0.00000 -0.00027 0.00000 0.00027 0.14489
Atom # 2 - L=2 ONLY - Spin component 2
1.15467 -0.02177 0.02672 -0.02177 0.00000 0.42925 -0.00885 0.01036 -0.00885 0.00000
-0.02177 1.15467 -0.01336 -0.02177 0.02314 -0.00885 0.42925 -0.00518 -0.00885 0.00897
0.02672 -0.01336 1.05928 -0.01336 0.00000 0.01208 -0.00604 0.39170 -0.00604 0.00000
-0.02177 -0.02177 -0.01336 1.15467 -0.02314 -0.00885 -0.00885 -0.00518 0.42925 -0.00897
0.00000 0.02314 0.00000 -0.02314 1.05928 0.00000 0.01046 0.00000 -0.01046 0.39170
0.42925 -0.00885 0.01208 -0.00885 0.00000 0.16415 -0.00363 0.00474 -0.00363 0.00000
-0.00885 0.42925 -0.00604 -0.00885 0.01046 -0.00363 0.16415 -0.00237 -0.00363 0.00410
0.01036 -0.00518 0.39170 -0.00518 0.00000 0.00474 -0.00237 0.15184 -0.00237 0.00000
-0.00885 -0.00885 -0.00604 0.42925 -0.01046 -0.00363 -0.00363 -0.00237 0.16415 -0.00410
0.00000 0.00897 0.00000 -0.00897 0.39170 0.00000 0.00410 0.00000 -0.00410 0.15184
Atom # 3 - L=1 ONLY - Spin component 1
1.06834 -0.00318 -0.00318 0.05053 -0.00033 -0.00033
-0.00318 1.06834 -0.00318 -0.00033 0.05053 -0.00033
-0.00318 -0.00318 1.06834 -0.00033 -0.00033 0.05053
0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
Atom # 3 - L=1 ONLY - Spin component 2
1.06837 -0.00319 -0.00319 0.05053 -0.00033 -0.00033
-0.00319 1.06837 -0.00319 -0.00033 0.05053 -0.00033
-0.00319 -0.00319 1.06837 -0.00033 -0.00033 0.05053
0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
Atom # 4 - L=1 ONLY - Spin component 1
1.06834 -0.00318 -0.00318 0.05053 -0.00033 -0.00033
-0.00318 1.06834 -0.00318 -0.00033 0.05053 -0.00033
-0.00318 -0.00318 1.06834 -0.00033 -0.00033 0.05053
0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
Atom # 4 - L=1 ONLY - Spin component 2
1.06837 -0.00319 -0.00319 0.05053 -0.00033 -0.00033
-0.00319 1.06837 -0.00319 -0.00033 0.05053 -0.00033
-0.00319 -0.00319 1.06837 -0.00033 -0.00033 0.05053
0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
---------- DFT+U DATA ---------------------------------------------------
====== For Atom 1, occupations for correlated orbitals. lpawu = 2
Atom 1. Occ. for lpawu and for spin 1 = 4.40560
Atom 1. Occ. for lpawu and for spin 2 = 4.22762
=> On atom 1, local Mag. for lpawu is -0.177977
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.91110 -0.01694 0.02066 -0.01694 0.00000
-0.01694 0.91110 -0.01033 -0.01694 0.01789
0.02066 -0.01033 0.83615 -0.01033 0.00000
-0.01694 -0.01694 -0.01033 0.91110 -0.01789
0.00000 0.01789 0.00000 -0.01789 0.83615
Occupation matrix for spin 2
0.87685 -0.01113 0.00062 -0.01113 0.00000
-0.01113 0.87685 -0.00031 -0.01113 0.00054
0.00062 -0.00031 0.79854 -0.00031 0.00000
-0.01113 -0.01113 -0.00031 0.87685 -0.00054
0.00000 0.00054 0.00000 -0.00054 0.79854
====== For Atom 2, occupations for correlated orbitals. lpawu = 2
Atom 2. Occ. for lpawu and for spin 1 = 4.23050
Atom 2. Occ. for lpawu and for spin 2 = 4.40815
=> On atom 2, local Mag. for lpawu is 0.177653
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.87734 -0.01108 0.00066 -0.01108 0.00000
-0.01108 0.87734 -0.00033 -0.01108 0.00057
0.00066 -0.00033 0.79923 -0.00033 0.00000
-0.01108 -0.01108 -0.00033 0.87734 -0.00057
0.00000 0.00057 0.00000 -0.00057 0.79923
Occupation matrix for spin 2
0.91147 -0.01696 0.02070 -0.01696 0.00000
-0.01696 0.91147 -0.01035 -0.01696 0.01793
0.02070 -0.01035 0.83688 -0.01035 0.00000
-0.01696 -0.01696 -0.01035 0.91147 -0.01793
0.00000 0.01793 0.00000 -0.01793 0.83688
====== For Atom 3, occupations for correlated orbitals. lpawu = 1
Atom 3. Occ. for lpawu and for spin 1 = 1.55467
Atom 3. Occ. for lpawu and for spin 2 = 1.55472
=> On atom 3, local Mag. for lpawu is 0.000050
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.51822 -0.00081 -0.00081
-0.00081 0.51822 -0.00081
-0.00081 -0.00081 0.51822
Occupation matrix for spin 2
0.51824 -0.00081 -0.00081
-0.00081 0.51824 -0.00081
-0.00081 -0.00081 0.51824
====== For Atom 4, occupations for correlated orbitals. lpawu = 1
Atom 4. Occ. for lpawu and for spin 1 = 1.55467
Atom 4. Occ. for lpawu and for spin 2 = 1.55472
=> On atom 4, local Mag. for lpawu is 0.000050
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.51822 -0.00081 -0.00081
-0.00081 0.51822 -0.00081
-0.00081 -0.00081 0.51822
Occupation matrix for spin 2
0.51824 -0.00081 -0.00081
-0.00081 0.51824 -0.00081
-0.00081 -0.00081 0.51824
---------- calculate U, (J) start ----------
All pawujat ok and equal to 1
macro_uj=1 and nspden=2: standard determination of Hubbard U-parameter
*********************************************************************
************************ Linear Response U ************************
Info printed for perturbed atom: 1
Perturbations Occupations
--------------- -----------------------------
alpha [eV] Unscreened Screened
--------------- -----------------------------
0.0000000000 8.6380182458 8.6380182458
0.0500000000 8.6192514500 8.6332174908
Scalar response functions:
Chi0 [eV^-1]: -0.83408
Chi [eV^-1]: -0.09602
The scalar U from the two-point regression scheme is 9.21610 eV.
*********************************************************************
*********************************************************************
Note: For more reliable linear regressions of the response
matrices, it is advised that you have more than two points.
See the LRUJ protocol for more information.
URES ii nat r_max U(J)[eV] U_ASA[eV] U_inf[eV]
URES 1 1 0.00000 2.30403 1.85413 1.79356
URES 2 8 11.14400 8.07831 6.50089 6.28853
URES 3 27 12.45940 8.87520 7.14217 6.90886
URES 4 64 22.28800 9.07213 7.30065 7.06216
URES 5 125 24.28780 9.14238 7.35718 7.11684
URES 6 216 33.43200 9.17344 7.38217 7.14102
---------- calculate U, (J) end --------------
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 32.750E-14; max= 53.822E-13
reduced coordinates (array xred) for 4 atoms
0.000000000000 0.000000000000 0.000000000000
0.500000000000 0.500000000000 0.500000000000
0.250000000000 0.250000000000 0.250000000000
0.750000000000 0.750000000000 0.750000000000
rms dE/dt= 1.4767E-03; max dE/dt= 2.0884E-03; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
2 0.000000000000 0.000000000000 0.000000000000
3 -0.002088362535 -0.002088362535 -0.002088362535
4 0.002088362535 0.002088362535 0.002088362535
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
2 4.16991641908672 4.16991641908672 4.16991641908672
3 2.08495820954336 2.08495820954336 2.08495820954336
4 6.25487462863008 6.25487462863008 6.25487462863008
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
2 -0.00000000000000 -0.00000000000000 -0.00000000000000
3 0.00013251031312 0.00013251031312 0.00013251031312
4 -0.00013251031312 -0.00013251031312 -0.00013251031312
frms,max,avg= 9.3698941E-05 1.3251031E-04 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 -0.00000000000000 -0.00000000000000
2 -0.00000000000000 -0.00000000000000 -0.00000000000000
3 0.00681395426725 0.00681395426725 0.00681395426725
4 -0.00681395426725 -0.00681395426725 -0.00681395426725
frms,max,avg= 4.8181933E-03 6.8139543E-03 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.880000000000 7.880000000000 7.880000000000 bohr
= 4.169916419087 4.169916419087 4.169916419087 angstroms
Fermi (or HOMO) energy (hartree) = 0.40850 Average Vxc (hartree)= -0.46912
Eigenvalues (hartree) for nkpt= 10 k points, SPIN UP:
kpt# 1, nband= 26, wtk= 0.03125, kpt= 0.1250 0.1250 0.1250 (reduced coord)
-4.14374 -4.13607 -2.37239 -2.33959 -2.33377 -2.33377 -2.31007 -2.31007
-0.40313 -0.35590 0.07034 0.21141 0.21141 0.22739 0.22739 0.25301
0.26198 0.26198 0.31991 0.31991 0.37558 0.38825 0.38825 0.39763
0.40073 0.43735
prteigrs : prtvol=0 or 1, do not print more k-points.
Eigenvalues (hartree) for nkpt= 10 k points, SPIN DOWN:
kpt# 1, nband= 26, wtk= 0.03125, kpt= 0.1250 0.1250 0.1250 (reduced coord)
-4.14136 -4.13845 -2.37212 -2.33984 -2.33314 -2.33314 -2.31069 -2.31069
-0.40313 -0.35590 0.07034 0.21141 0.21141 0.22739 0.22739 0.25301
0.26197 0.26197 0.31991 0.31991 0.37556 0.38825 0.38825 0.39763
0.40077 0.43736
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 4, }
comment : Components of total free energy in Hartree
kinetic : 8.94669717817659E+01
hartree : 8.65267496852782E+01
xc : -2.18476794835206E+01
Ewald energy : -2.39331554544165E+02
psp_core : 1.04313020493122E+01
local_psp : -2.68557569776284E+02
PAW spherical_terms : -2.03990180084070E+01
total_energy : -3.63710798296021E+02
total_energy_eV : -9.89707501426712E+03
...
--- !EnergyTermsDC
iteration_state : {dtset: 4, }
comment : '"Double-counting" decomposition of free energy'
band_energy : -4.00328214044425E+01
Ewald energy : -2.39331554544165E+02
psp_core : 1.04313020493122E+01
xc_dc : -7.99671660270088E+01
PAW spherical_terms : -1.47947012791744E+01
total_energy_dc : -3.63694941205479E+02
total_energy_dc_eV : -9.89664352085163E+03
...
--- !EnergyTermsWithPAWCore
iteration_state : {dtset: 4, }
comment : 'Components of total free energy, including PAW core contributions'
Total energy : -3.18179009074010E+03
Total energy DC : -3.18177423364956E+03
Total energy (eV) : -8.65809191127639E+04
Total energy DC (eV): -8.65804876193484E+04
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42464714E-01 sigma(3 2)= -4.89478733E-04
sigma(2 2)= 1.42464714E-01 sigma(3 1)= -4.89478733E-04
sigma(3 3)= 1.42464714E-01 sigma(2 1)= -4.89478733E-04
-Cartesian components of stress tensor (GPa) [Pressure= -4.1915E+03 GPa]
- sigma(1 1)= 4.19145659E+03 sigma(3 2)= -1.44009615E+01
- sigma(2 2)= 4.19145659E+03 sigma(3 1)= -1.44009615E+01
- sigma(3 3)= 4.19145659E+03 sigma(2 1)= -1.44009615E+01
================================================================================
== DATASET 5 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 5, }
dimensions: {natom: 4, nkpt: 10, mband: 26, nsppol: 2, nspinor: 1, nspden: 2, mpw: 376, }
cutoff_energies: {ecut: 10.0, pawecutdg: 20.0, }
electrons: {nelect: 4.80000000E+01, charge: 0.00000000E+00, occopt: 0.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 0, ionmov: 0, optcell: 0, iscf: 17, paral_kgb: 0, }
...
mkfilename: getwfk from: ./tlruj_1.o_WFK
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 3.9400000 3.9400000 7.8800000 G(1)= -0.0634518 -0.0634518 0.1903553
R(2)= 7.8800000 3.9400000 3.9400000 G(2)= 0.1903553 -0.0634518 -0.0634518
R(3)= 3.9400000 7.8800000 3.9400000 G(3)= -0.0634518 0.1903553 -0.0634518
Unit cell volume ucvol= 2.4465194E+02 bohr^3
Angles (23,13,12)= 3.35573098E+01 3.35573098E+01 3.35573098E+01 degrees
Coarse grid specifications (used for wave-functions):
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 30 30 30
ecut(hartree)= 10.000 => boxcut(ratio)= 2.18911
Fine grid specifications (used for densities):
getcut: wavevector= 0.0000 0.0000 0.0000 ngfft= 40 40 40
ecut(hartree)= 20.000 => boxcut(ratio)= 2.06006
--------------------------------------------------------------------------------
-inwffil : will read wavefunctions from disk file ./tlruj_1.o_WFK
_setup2: Arith. and geom. avg. npw (full set) are 368.875 368.830
******************************************
DFT+U Method used: FLL
******************************************
================================================================================
--- !BeginCycle
iteration_state: {dtset: 5, }
solver: {iscf: 17, nstep: 30, nline: 4, wfoptalg: 10, }
tolerances: {tolvrs: 1.00E-09, }
...
iter Etot(hartree) deltaE(h) residm nres2 difmagsph maxmagsph
ETOT 1 -363.67922154708 -3.637E+02 5.914E-08 2.311E-01 1.524E-01 1.524E-01
ETOT 2 -363.68213346012 -2.912E-03 5.813E-07 4.661E+00 8.463E-02 8.109E-02
ETOT 3 -363.67908299064 3.050E-03 1.930E-04 1.244E-02 8.597E-02 1.538E-01
ETOT 4 -363.67908195516 1.035E-06 5.630E-07 6.073E-04 1.256E-03 1.550E-01
ETOT 5 -363.67908203977 -8.461E-08 3.281E-07 1.308E-04 2.456E-04 1.553E-01
ETOT 6 -363.67908229263 -2.529E-07 2.570E-08 1.486E-04 5.307E-04 1.558E-01
ETOT 7 -363.67908228016 1.248E-08 1.661E-08 3.523E-05 3.568E-04 1.562E-01
ETOT 8 -363.67908229293 -1.278E-08 2.671E-09 3.097E-05 1.001E-04 1.563E-01
ETOT 9 -363.67908231742 -2.449E-08 2.987E-09 2.349E-05 2.421E-04 1.565E-01
ETOT 10 -363.67908238817 -7.075E-08 5.234E-09 2.201E-05 7.100E-04 1.572E-01
ETOT 11 -363.67908242230 -3.413E-08 1.977E-08 1.513E-06 1.316E-03 1.585E-01
ETOT 12 -363.67908243998 -1.769E-08 1.259E-09 2.766E-07 1.839E-04 1.587E-01
ETOT 13 -363.67908244334 -3.360E-09 1.427E-09 1.039E-07 1.404E-04 1.588E-01
ETOT 14 -363.67908244566 -2.320E-09 2.805E-10 1.292E-08 4.331E-05 1.589E-01
ETOT 15 -363.67908244583 -1.701E-10 2.329E-10 5.457E-09 9.291E-06 1.589E-01
ETOT 16 -363.67908244591 -7.213E-11 5.206E-11 1.050E-09 2.303E-05 1.589E-01
ETOT 17 -363.67908244591 -6.196E-12 3.529E-11 6.782E-10 1.946E-06 1.589E-01
At SCF step 17 nres2 = 6.78E-10 < tolvrs= 1.00E-09 =>converged.
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42458350E-01 sigma(3 2)= -4.91229278E-04
sigma(2 2)= 1.42458350E-01 sigma(3 1)= -4.91229278E-04
sigma(3 3)= 1.42458350E-01 sigma(2 1)= -4.91229278E-04
--- !ResultsGS
iteration_state: {dtset: 5, }
comment : Summary of ground state results
lattice_vectors:
- [ 3.9400000, 3.9400000, 7.8800000, ]
- [ 7.8800000, 3.9400000, 3.9400000, ]
- [ 3.9400000, 7.8800000, 3.9400000, ]
lattice_lengths: [ 9.65099, 9.65099, 9.65099, ]
lattice_angles: [ 33.557, 33.557, 33.557, ] # degrees, (23, 13, 12)
lattice_volume: 2.4465194E+02
convergence: {deltae: -6.196E-12, res2: 6.782E-10, residm: 3.529E-11, diffor: null, }
etotal : -3.63679082E+02
entropy : 0.00000000E+00
fermie : 4.08931266E-01
cartesian_stress_tensor: # hartree/bohr^3
- [ 1.42458350E-01, -4.91229278E-04, -4.91229278E-04, ]
- [ -4.91229278E-04, 1.42458350E-01, -4.91229278E-04, ]
- [ -4.91229278E-04, -4.91229278E-04, 1.42458350E-01, ]
pressure_GPa: -4.1913E+03
xred :
- [ 0.0000E+00, 0.0000E+00, 0.0000E+00, Ni]
- [ 5.0000E-01, 5.0000E-01, 5.0000E-01, Ni]
- [ 2.5000E-01, 2.5000E-01, 2.5000E-01, O]
- [ 7.5000E-01, 7.5000E-01, 7.5000E-01, O]
cartesian_forces: # hartree/bohr
- [ -2.71050543E-20, 2.71050543E-20, -0.00000000E+00, ]
- [ -2.71050543E-20, 2.71050543E-20, -0.00000000E+00, ]
- [ 2.65775992E-04, 2.65775992E-04, 2.65775992E-04, ]
- [ -2.65775992E-04, -2.65775992E-04, -2.65775992E-04, ]
force_length_stats: {min: 3.83323354E-20, max: 4.60337521E-04, mean: 2.30168761E-04, }
...
Integrated electronic and magnetization densities in atomic spheres:
------------------------------------------------------------------------------------------------------------
Radius=ratsph(iatom), smearing ratsm= 0.0000. Diff(up-dn)=approximate z local magnetic moment.
Atom Radius up_density dn_density Total(up+dn) Diff(up-dn)
1 Ni 1.81432 8.021144 7.862221 15.883365 0.158923
2 Ni 1.81432 7.866357 8.024650 15.891007 -0.158293
3 O 1.41465 2.292341 2.292552 4.584894 -0.000211
4 O 1.41465 2.292341 2.292552 4.584894 -0.000211
------------------------------------------------------------------------------------------------------------
Sum: 20.472184 20.471975 40.944159 0.000208
Total magnetization (from the atomic spheres): 0.000208
Total magnetization (exact up - dn): 0.000000
PAW TEST:
==== Compensation charge inside spheres ============
The following values must be close to each other ...
Compensation charge over spherical meshes = 12.716914199288876
Compensation charge over fine fft grid = 12.717201808787767
==== Results concerning PAW augmentation regions ====
Total pseudopotential strength Dij (hartree):
Atom # 1 - Spin component 1
-2.33345 0.37622 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00172 -0.00172 0.00000 -0.00172 ...
0.37622 0.04433 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00034 0.00034 0.00000 0.00034 ...
0.00000 0.00000 -2.24455 -0.00127 -0.00127 0.82343 0.00078 0.00078 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00127 -2.24455 -0.00127 0.00078 0.82343 0.00078 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00127 -0.00127 -2.24455 0.00078 0.00078 0.82343 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.82343 0.00078 0.00078 0.36934 -0.00047 -0.00047 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00078 0.82343 0.00078 -0.00047 0.36934 -0.00047 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00078 0.00078 0.82343 -0.00047 -0.00047 0.36934 0.00000 0.00000 0.00000 0.00000 ...
-0.00172 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.91037 -0.00103 0.00094 -0.00103 ...
-0.00172 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00103 -0.91037 -0.00047 -0.00103 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00094 -0.00047 -0.91166 -0.00047 ...
-0.00172 0.00034 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00103 -0.00103 -0.00047 -0.91037 ...
... only 12 components have been written...
Atom # 1 - Spin component 2
-2.32960 0.37551 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00223 -0.00223 0.00000 -0.00223 ...
0.37551 0.04443 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00044 0.00044 0.00000 0.00044 ...
0.00000 0.00000 -2.24132 -0.00165 -0.00165 0.82163 0.00099 0.00099 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00165 -2.24132 -0.00165 0.00099 0.82163 0.00099 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 -0.00165 -0.00165 -2.24132 0.00099 0.00099 0.82163 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.82163 0.00099 0.00099 0.37042 -0.00060 -0.00060 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00099 0.82163 0.00099 -0.00060 0.37042 -0.00060 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00099 0.00099 0.82163 -0.00060 -0.00060 0.37042 0.00000 0.00000 0.00000 0.00000 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.90758 -0.00114 0.00137 -0.00114 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00114 -0.90758 -0.00069 -0.00114 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00137 -0.00069 -0.90867 -0.00069 ...
-0.00223 0.00044 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00114 -0.00114 -0.00069 -0.90758 ...
... only 12 components have been written...
Atom # 4 - Spin component 1
0.61335 -1.75544 0.00002 0.00002 0.00002 -0.00011 -0.00011 -0.00011
-1.75544 5.08470 -0.00004 -0.00004 -0.00004 0.00029 0.00029 0.00029
0.00002 -0.00004 -0.29392 -0.00002 -0.00002 0.88790 0.00011 0.00011
0.00002 -0.00004 -0.00002 -0.29392 -0.00002 0.00011 0.88790 0.00011
0.00002 -0.00004 -0.00002 -0.00002 -0.29392 0.00011 0.00011 0.88790
-0.00011 0.00029 0.88790 0.00011 0.00011 0.12123 -0.00056 -0.00056
-0.00011 0.00029 0.00011 0.88790 0.00011 -0.00056 0.12123 -0.00056
-0.00011 0.00029 0.00011 0.00011 0.88790 -0.00056 -0.00056 0.12123
Atom # 4 - Spin component 2
0.61335 -1.75547 -0.00001 -0.00001 -0.00001 0.00006 0.00006 0.00006
-1.75547 5.08478 0.00003 0.00003 0.00003 -0.00017 -0.00017 -0.00017
-0.00001 0.00003 -0.29392 -0.00002 -0.00002 0.88792 0.00011 0.00011
-0.00001 0.00003 -0.00002 -0.29392 -0.00002 0.00011 0.88792 0.00011
-0.00001 0.00003 -0.00002 -0.00002 -0.29392 0.00011 0.00011 0.88792
0.00006 -0.00017 0.88792 0.00011 0.00011 0.12112 -0.00055 -0.00055
0.00006 -0.00017 0.00011 0.88792 0.00011 -0.00055 0.12112 -0.00055
0.00006 -0.00017 0.00011 0.00011 0.88792 -0.00055 -0.00055 0.12112
Augmentation waves occupancies Rhoij:
Atom # 1 - Spin component 1
0.97687 -0.16719 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00017 -0.00017 0.00000 -0.00017 ...
-0.16719 0.46268 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00442 -0.00442 0.00000 -0.00442 ...
0.00000 0.00000 1.06707 0.00002 0.00002 0.13560 0.00007 0.00007 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00002 1.06707 0.00002 0.00007 0.13560 0.00007 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00002 0.00002 1.06707 0.00007 0.00007 0.13560 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.13560 0.00007 0.00007 0.02463 0.00002 0.00002 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00007 0.13560 0.00007 0.00002 0.02463 0.00002 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00007 0.00007 0.13560 0.00002 0.00002 0.02463 0.00000 0.00000 0.00000 0.00000 ...
-0.00017 -0.00442 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 1.15379 -0.02175 0.02667 -0.02175 ...
-0.00017 -0.00442 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.02175 1.15379 -0.01334 -0.02175 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.02667 -0.01334 1.05774 -0.01334 ...
-0.00017 -0.00442 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.02175 -0.02175 -0.01334 1.15379 ...
... only 12 components have been written...
Atom # 1 - Spin component 2
0.97680 -0.16763 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00009 -0.00009 0.00000 -0.00009 ...
-0.16763 0.46353 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.00011 -0.00011 0.00000 -0.00011 ...
0.00000 0.00000 1.06704 0.00003 0.00003 0.13549 0.00012 0.00012 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00003 1.06704 0.00003 0.00012 0.13549 0.00012 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00003 0.00003 1.06704 0.00012 0.00012 0.13549 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.13549 0.00012 0.00012 0.02455 0.00004 0.00004 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00012 0.13549 0.00012 0.00004 0.02455 0.00004 0.00000 0.00000 0.00000 0.00000 ...
0.00000 0.00000 0.00012 0.00012 0.13549 0.00004 0.00004 0.02455 0.00000 0.00000 0.00000 0.00000 ...
-0.00009 -0.00011 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 1.10853 -0.01449 0.00051 -0.01449 ...
-0.00009 -0.00011 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.01449 1.10853 -0.00026 -0.01449 ...
0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00051 -0.00026 1.00973 -0.00026 ...
-0.00009 -0.00011 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -0.01449 -0.01449 -0.00026 1.10853 ...
... only 12 components have been written...
Atom # 4 - Spin component 1
1.01162 0.01395 -0.00072 -0.00072 -0.00072 0.00003 0.00003 0.00003
0.01395 0.00120 0.00011 0.00011 0.00011 -0.00000 -0.00000 -0.00000
-0.00072 0.00011 1.06836 -0.00317 -0.00317 0.05053 -0.00033 -0.00033
-0.00072 0.00011 -0.00317 1.06836 -0.00317 -0.00033 0.05053 -0.00033
-0.00072 0.00011 -0.00317 -0.00317 1.06836 -0.00033 -0.00033 0.05053
0.00003 -0.00000 0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
0.00003 -0.00000 -0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
0.00003 -0.00000 -0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
Atom # 4 - Spin component 2
1.01169 0.01394 0.00092 0.00092 0.00092 -0.00003 -0.00003 -0.00003
0.01394 0.00120 -0.00017 -0.00017 -0.00017 -0.00000 -0.00000 -0.00000
0.00092 -0.00017 1.06841 -0.00318 -0.00318 0.05053 -0.00033 -0.00033
0.00092 -0.00017 -0.00318 1.06841 -0.00318 -0.00033 0.05053 -0.00033
0.00092 -0.00017 -0.00318 -0.00318 1.06841 -0.00033 -0.00033 0.05053
-0.00003 -0.00000 0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00003 -0.00000 -0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
-0.00003 -0.00000 -0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
"PAW+U" part of augmentation waves occupancies Rhoij:
Atom # 1 - L=2 ONLY - Spin component 1
1.15379 -0.02175 0.02667 -0.02175 0.00000 0.42883 -0.00885 0.01032 -0.00885 0.00000
-0.02175 1.15379 -0.01334 -0.02175 0.02310 -0.00885 0.42883 -0.00516 -0.00885 0.00894
0.02667 -0.01334 1.05774 -0.01334 0.00000 0.01204 -0.00602 0.39104 -0.00602 0.00000
-0.02175 -0.02175 -0.01334 1.15379 -0.02310 -0.00885 -0.00885 -0.00516 0.42883 -0.00894
0.00000 0.02310 0.00000 -0.02310 1.05774 0.00000 0.01043 0.00000 -0.01043 0.39104
0.42883 -0.00885 0.01204 -0.00885 0.00000 0.16397 -0.00363 0.00472 -0.00363 0.00000
-0.00885 0.42883 -0.00602 -0.00885 0.01043 -0.00363 0.16397 -0.00236 -0.00363 0.00408
0.01032 -0.00516 0.39104 -0.00516 0.00000 0.00472 -0.00236 0.15157 -0.00236 0.00000
-0.00885 -0.00885 -0.00602 0.42883 -0.01043 -0.00363 -0.00363 -0.00236 0.16397 -0.00408
0.00000 0.00894 0.00000 -0.00894 0.39104 0.00000 0.00408 0.00000 -0.00408 0.15157
Atom # 1 - L=2 ONLY - Spin component 2
1.10853 -0.01449 0.00051 -0.01449 0.00000 0.40813 -0.00572 -0.00029 -0.00572 0.00000
-0.01449 1.10853 -0.00026 -0.01449 0.00044 -0.00572 0.40813 0.00014 -0.00572 -0.00025
0.00051 -0.00026 1.00973 -0.00026 0.00000 -0.00051 0.00025 0.37281 0.00025 0.00000
-0.01449 -0.01449 -0.00026 1.10853 -0.00044 -0.00572 -0.00572 0.00014 0.40813 0.00025
0.00000 0.00044 0.00000 -0.00044 1.00973 0.00000 -0.00044 0.00000 0.00044 0.37281
0.40813 -0.00572 -0.00051 -0.00572 0.00000 0.15448 -0.00229 -0.00035 -0.00229 0.00000
-0.00572 0.40813 0.00025 -0.00572 -0.00044 -0.00229 0.15448 0.00018 -0.00229 -0.00031
-0.00029 0.00014 0.37281 0.00014 0.00000 -0.00035 0.00018 0.14461 0.00018 0.00000
-0.00572 -0.00572 0.00025 0.40813 0.00044 -0.00229 -0.00229 0.00018 0.15448 0.00031
0.00000 -0.00025 0.00000 0.00025 0.37281 0.00000 -0.00031 0.00000 0.00031 0.14461
Atom # 2 - L=2 ONLY - Spin component 1
1.10991 -0.01431 0.00063 -0.01431 0.00000 0.40882 -0.00564 -0.00021 -0.00564 0.00000
-0.01431 1.10991 -0.00031 -0.01431 0.00054 -0.00564 0.40882 0.00011 -0.00564 -0.00018
0.00063 -0.00031 1.01152 -0.00031 0.00000 -0.00040 0.00020 0.37356 0.00020 0.00000
-0.01431 -0.01431 -0.00031 1.10991 -0.00054 -0.00564 -0.00564 0.00011 0.40882 0.00018
0.00000 0.00054 0.00000 -0.00054 1.01152 0.00000 -0.00035 0.00000 0.00035 0.37356
0.40882 -0.00564 -0.00040 -0.00564 0.00000 0.15481 -0.00225 -0.00030 -0.00225 0.00000
-0.00564 0.40882 0.00020 -0.00564 -0.00035 -0.00225 0.15481 0.00015 -0.00225 -0.00026
-0.00021 0.00011 0.37356 0.00011 0.00000 -0.00030 0.00015 0.14490 0.00015 0.00000
-0.00564 -0.00564 0.00020 0.40882 0.00035 -0.00225 -0.00225 0.00015 0.15481 0.00026
0.00000 -0.00018 0.00000 0.00018 0.37356 0.00000 -0.00026 0.00000 0.00026 0.14490
Atom # 2 - L=2 ONLY - Spin component 2
1.15474 -0.02181 0.02679 -0.02181 0.00000 0.42930 -0.00887 0.01039 -0.00887 0.00000
-0.02181 1.15474 -0.01340 -0.02181 0.02320 -0.00887 0.42930 -0.00520 -0.00887 0.00900
0.02679 -0.01340 1.05960 -0.01340 0.00000 0.01213 -0.00606 0.39182 -0.00606 0.00000
-0.02181 -0.02181 -0.01340 1.15474 -0.02320 -0.00887 -0.00887 -0.00520 0.42930 -0.00900
0.00000 0.02320 0.00000 -0.02320 1.05960 0.00000 0.01050 0.00000 -0.01050 0.39182
0.42930 -0.00887 0.01213 -0.00887 0.00000 0.16418 -0.00364 0.00476 -0.00364 0.00000
-0.00887 0.42930 -0.00606 -0.00887 0.01050 -0.00364 0.16418 -0.00238 -0.00364 0.00412
0.01039 -0.00520 0.39182 -0.00520 0.00000 0.00476 -0.00238 0.15188 -0.00238 0.00000
-0.00887 -0.00887 -0.00606 0.42930 -0.01050 -0.00364 -0.00364 -0.00238 0.16418 -0.00412
0.00000 0.00900 0.00000 -0.00900 0.39182 0.00000 0.00412 0.00000 -0.00412 0.15188
Atom # 3 - L=1 ONLY - Spin component 1
1.06836 -0.00317 -0.00317 0.05053 -0.00033 -0.00033
-0.00317 1.06836 -0.00317 -0.00033 0.05053 -0.00033
-0.00317 -0.00317 1.06836 -0.00033 -0.00033 0.05053
0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
Atom # 3 - L=1 ONLY - Spin component 2
1.06841 -0.00318 -0.00318 0.05053 -0.00033 -0.00033
-0.00318 1.06841 -0.00318 -0.00033 0.05053 -0.00033
-0.00318 -0.00318 1.06841 -0.00033 -0.00033 0.05053
0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
Atom # 4 - L=1 ONLY - Spin component 1
1.06836 -0.00317 -0.00317 0.05053 -0.00033 -0.00033
-0.00317 1.06836 -0.00317 -0.00033 0.05053 -0.00033
-0.00317 -0.00317 1.06836 -0.00033 -0.00033 0.05053
0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
Atom # 4 - L=1 ONLY - Spin component 2
1.06841 -0.00318 -0.00318 0.05053 -0.00033 -0.00033
-0.00318 1.06841 -0.00318 -0.00033 0.05053 -0.00033
-0.00318 -0.00318 1.06841 -0.00033 -0.00033 0.05053
0.05053 -0.00033 -0.00033 0.00287 -0.00001 -0.00001
-0.00033 0.05053 -0.00033 -0.00001 0.00287 -0.00001
-0.00033 -0.00033 0.05053 -0.00001 -0.00001 0.00287
---------- DFT+U DATA ---------------------------------------------------
====== For Atom 1, occupations for correlated orbitals. lpawu = 2
Atom 1. Occ. for lpawu and for spin 1 = 4.40378
Atom 1. Occ. for lpawu and for spin 2 = 4.22453
=> On atom 1, local Mag. for lpawu is -0.179246
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.91080 -0.01694 0.02067 -0.01694 0.00000
-0.01694 0.91080 -0.01034 -0.01694 0.01790
0.02067 -0.01034 0.83569 -0.01034 0.00000
-0.01694 -0.01694 -0.01034 0.91080 -0.01790
0.00000 0.01790 0.00000 -0.01790 0.83569
Occupation matrix for spin 2
0.87624 -0.01134 0.00058 -0.01134 0.00000
-0.01134 0.87624 -0.00029 -0.01134 0.00050
0.00058 -0.00029 0.79791 -0.00029 0.00000
-0.01134 -0.01134 -0.00029 0.87624 -0.00050
0.00000 0.00050 0.00000 -0.00050 0.79791
====== For Atom 2, occupations for correlated orbitals. lpawu = 2
Atom 2. Occ. for lpawu and for spin 1 = 4.23040
Atom 2. Occ. for lpawu and for spin 2 = 4.40879
=> On atom 2, local Mag. for lpawu is 0.178393
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.87727 -0.01120 0.00066 -0.01120 0.00000
-0.01120 0.87727 -0.00033 -0.01120 0.00057
0.00066 -0.00033 0.79929 -0.00033 0.00000
-0.01120 -0.01120 -0.00033 0.87727 -0.00057
0.00000 0.00057 0.00000 -0.00057 0.79929
Occupation matrix for spin 2
0.91151 -0.01699 0.02076 -0.01699 0.00000
-0.01699 0.91151 -0.01038 -0.01699 0.01798
0.02076 -0.01038 0.83713 -0.01038 0.00000
-0.01699 -0.01699 -0.01038 0.91151 -0.01798
0.00000 0.01798 0.00000 -0.01798 0.83713
====== For Atom 3, occupations for correlated orbitals. lpawu = 1
Atom 3. Occ. for lpawu and for spin 1 = 1.55464
Atom 3. Occ. for lpawu and for spin 2 = 1.55475
=> On atom 3, local Mag. for lpawu is 0.000105
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.51821 -0.00080 -0.00080
-0.00080 0.51821 -0.00080
-0.00080 -0.00080 0.51821
Occupation matrix for spin 2
0.51825 -0.00081 -0.00081
-0.00081 0.51825 -0.00081
-0.00081 -0.00081 0.51825
====== For Atom 4, occupations for correlated orbitals. lpawu = 1
Atom 4. Occ. for lpawu and for spin 1 = 1.55464
Atom 4. Occ. for lpawu and for spin 2 = 1.55475
=> On atom 4, local Mag. for lpawu is 0.000105
== Occupation matrix for correlated orbitals:
Occupation matrix for spin 1
0.51821 -0.00080 -0.00080
-0.00080 0.51821 -0.00080
-0.00080 -0.00080 0.51821
Occupation matrix for spin 2
0.51825 -0.00081 -0.00081
-0.00081 0.51825 -0.00081
-0.00081 -0.00081 0.51825
---------- calculate U, (J) start ----------
All pawujat ok and equal to 1
macro_uj=1 and nspden=2: standard determination of Hubbard U-parameter
*********************************************************************
************************ Linear Response U ************************
Info printed for perturbed atom: 1
Perturbations Occupations
--------------- -----------------------------
alpha [eV] Unscreened Screened
--------------- -----------------------------
0.0000000000 8.6380182458 8.6380182458
0.1000000000 8.5980252049 8.6283148758
Scalar response functions:
Chi0 [eV^-1]: -0.88873
Chi [eV^-1]: -0.09703
The scalar U from the two-point regression scheme is 9.18050 eV.
*********************************************************************
*********************************************************************
Note: For more reliable linear regressions of the response
matrices, it is advised that you have more than two points.
See the LRUJ protocol for more information.
URES ii nat r_max U(J)[eV] U_ASA[eV] U_inf[eV]
URES 1 1 0.00000 2.29513 1.84696 1.78663
URES 2 8 11.14400 8.04711 6.47578 6.26423
URES 3 27 12.45940 8.84092 7.11458 6.88217
URES 4 64 22.28800 9.03709 7.27245 7.03488
URES 5 125 24.28780 9.10706 7.32876 7.08935
URES 6 216 33.43200 9.13800 7.35365 7.11344
---------- calculate U, (J) end --------------
================================================================================
----iterations are completed or convergence reached----
Mean square residual over all n,k,spin= 85.849E-15; max= 35.289E-12
reduced coordinates (array xred) for 4 atoms
0.000000000000 0.000000000000 0.000000000000
0.500000000000 0.500000000000 0.500000000000
0.250000000000 0.250000000000 0.250000000000
0.750000000000 0.750000000000 0.750000000000
rms dE/dt= 2.9618E-03; max dE/dt= 4.1886E-03; dE/dt below (all hartree)
1 0.000000000000 0.000000000000 0.000000000000
2 0.000000000000 0.000000000000 0.000000000000
3 -0.004188629632 -0.004188629632 -0.004188629632
4 0.004188629632 0.004188629632 0.004188629632
cartesian coordinates (angstrom) at end:
1 0.00000000000000 0.00000000000000 0.00000000000000
2 4.16991641908672 4.16991641908672 4.16991641908672
3 2.08495820954336 2.08495820954336 2.08495820954336
4 6.25487462863008 6.25487462863008 6.25487462863008
cartesian forces (hartree/bohr) at end:
1 -0.00000000000000 0.00000000000000 -0.00000000000000
2 -0.00000000000000 0.00000000000000 -0.00000000000000
3 0.00026577599186 0.00026577599186 0.00026577599186
4 -0.00026577599186 -0.00026577599186 -0.00026577599186
frms,max,avg= 1.8793201E-04 2.6577599E-04 0.000E+00 0.000E+00 0.000E+00 h/b
cartesian forces (eV/Angstrom) at end:
1 -0.00000000000000 0.00000000000000 -0.00000000000000
2 -0.00000000000000 0.00000000000000 -0.00000000000000
3 0.01366675099628 0.01366675099628 0.01366675099628
4 -0.01366675099628 -0.01366675099628 -0.01366675099628
frms,max,avg= 9.6638523E-03 1.3666751E-02 0.000E+00 0.000E+00 0.000E+00 e/A
length scales= 7.880000000000 7.880000000000 7.880000000000 bohr
= 4.169916419087 4.169916419087 4.169916419087 angstroms
Fermi (or HOMO) energy (hartree) = 0.40893 Average Vxc (hartree)= -0.46916
Eigenvalues (hartree) for nkpt= 10 k points, SPIN UP:
kpt# 1, nband= 26, wtk= 0.03125, kpt= 0.1250 0.1250 0.1250 (reduced coord)
-4.14570 -4.13562 -2.37334 -2.34006 -2.33491 -2.33491 -2.31028 -2.31028
-0.40302 -0.35577 0.07041 0.21163 0.21163 0.22778 0.22778 0.25312
0.26215 0.26215 0.32030 0.32030 0.37597 0.38867 0.38867 0.39799
0.40075 0.43774
prteigrs : prtvol=0 or 1, do not print more k-points.
Eigenvalues (hartree) for nkpt= 10 k points, SPIN DOWN:
kpt# 1, nband= 26, wtk= 0.03125, kpt= 0.1250 0.1250 0.1250 (reduced coord)
-4.14094 -4.14036 -2.37280 -2.34056 -2.33366 -2.33366 -2.31151 -2.31151
-0.40303 -0.35578 0.07040 0.21162 0.21162 0.22778 0.22778 0.25311
0.26214 0.26214 0.32031 0.32031 0.37594 0.38867 0.38867 0.39799
0.40082 0.43777
prteigrs : prtvol=0 or 1, do not print more k-points.
--- !EnergyTerms
iteration_state : {dtset: 5, }
comment : Components of total free energy in Hartree
kinetic : 8.94590398984871E+01
hartree : 8.65097584049733E+01
xc : -2.18469212836971E+01
Ewald energy : -2.39331554544165E+02
psp_core : 1.04313020493122E+01
local_psp : -2.68533458851143E+02
PAW spherical_terms : -2.03989579211862E+01
total_energy : -3.63710792247419E+02
total_energy_eV : -9.89707484967626E+03
...
--- !EnergyTermsDC
iteration_state : {dtset: 5, }
comment : '"Double-counting" decomposition of free energy'
band_energy : -4.00354704413136E+01
Ewald energy : -2.39331554544165E+02
psp_core : 1.04313020493122E+01
xc_dc : -7.99503807992052E+01
PAW spherical_terms : -1.47929787105398E+01
total_energy_dc : -3.63679082445912E+02
total_energy_dc_eV : -9.89621198201967E+03
...
--- !EnergyTermsWithPAWCore
iteration_state : {dtset: 5, }
comment : 'Components of total free energy, including PAW core contributions'
Total energy : -3.18179008469150E+03
Total energy DC : -3.18175837488999E+03
Total energy (eV) : -8.65809189481730E+04
Total energy DC (eV): -8.65800560805164E+04
...
Cartesian components of stress tensor (hartree/bohr^3)
sigma(1 1)= 1.42458350E-01 sigma(3 2)= -4.91229278E-04
sigma(2 2)= 1.42458350E-01 sigma(3 1)= -4.91229278E-04
sigma(3 3)= 1.42458350E-01 sigma(2 1)= -4.91229278E-04
-Cartesian components of stress tensor (GPa) [Pressure= -4.1913E+03 GPa]
- sigma(1 1)= 4.19126936E+03 sigma(3 2)= -1.44524643E+01
- sigma(2 2)= 4.19126936E+03 sigma(3 1)= -1.44524643E+01
- sigma(3 3)= 4.19126936E+03 sigma(2 1)= -1.44524643E+01
== END DATASET(S) ==============================================================
================================================================================
-outvars: echo values of variables after computation --------
acell 7.8800000000E+00 7.8800000000E+00 7.8800000000E+00 Bohr
amu 5.86900000E+01 5.86900000E+01 1.59994000E+01
atvshift1 -0.00551 -0.00551 -0.00551 -0.00551 -0.00551
-0.00551 -0.00551 -0.00551 -0.00551 -0.00551
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
atvshift2 -0.00367 -0.00367 -0.00367 -0.00367 -0.00367
-0.00367 -0.00367 -0.00367 -0.00367 -0.00367
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
atvshift3 -0.00184 -0.00184 -0.00184 -0.00184 -0.00184
-0.00184 -0.00184 -0.00184 -0.00184 -0.00184
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
atvshift4 0.00184 0.00184 0.00184 0.00184 0.00184
0.00184 0.00184 0.00184 0.00184 0.00184
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
atvshift5 0.00367 0.00367 0.00367 0.00367 0.00367
0.00367 0.00367 0.00367 0.00367 0.00367
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
0.00000 0.00000 0.00000 0.00000 0.00000
chkprim 0
chksymbreak 0
diemix 4.50000000E-01
dmatpuopt 3
ecut 1.00000000E+01 Hartree
etotal1 -3.6375846399E+02
etotal2 -3.6374257036E+02
etotal3 -3.6372668530E+02
etotal4 -3.6369494121E+02
etotal5 -3.6367908245E+02
fcart1 5.4210108624E-20 -6.7762635780E-20 -5.4210108624E-20
5.4210108624E-20 -6.7762635780E-20 -5.4210108624E-20
-3.8419124296E-04 -3.8419124296E-04 -3.8419124296E-04
3.8419124296E-04 3.8419124296E-04 3.8419124296E-04
fcart2 -0.0000000000E+00 2.7105054312E-20 -5.4210108624E-20
-0.0000000000E+00 2.7105054312E-20 -5.4210108624E-20
-2.5727030544E-04 -2.5727030544E-04 -2.5727030544E-04
2.5727030544E-04 2.5727030544E-04 2.5727030544E-04
fcart3 6.7762635780E-21 -2.0328790734E-20 6.7762635780E-21
6.7762635780E-21 -2.0328790734E-20 6.7762635780E-21
-1.3023297894E-04 -1.3023297894E-04 -1.3023297894E-04
1.3023297894E-04 1.3023297894E-04 1.3023297894E-04
fcart4 -0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
-0.0000000000E+00 -0.0000000000E+00 -0.0000000000E+00
1.3251031312E-04 1.3251031312E-04 1.3251031312E-04
-1.3251031312E-04 -1.3251031312E-04 -1.3251031312E-04
fcart5 -2.7105054312E-20 2.7105054312E-20 -0.0000000000E+00
-2.7105054312E-20 2.7105054312E-20 -0.0000000000E+00
2.6577599186E-04 2.6577599186E-04 2.6577599186E-04
-2.6577599186E-04 -2.6577599186E-04 -2.6577599186E-04
- fftalg 512
ixc -101130
jdtset 1 2 3 4 5
kpt 1.25000000E-01 1.25000000E-01 1.25000000E-01
3.75000000E-01 1.25000000E-01 1.25000000E-01
-3.75000000E-01 1.25000000E-01 1.25000000E-01
-1.25000000E-01 1.25000000E-01 1.25000000E-01
3.75000000E-01 3.75000000E-01 1.25000000E-01
-3.75000000E-01 3.75000000E-01 1.25000000E-01
-1.25000000E-01 3.75000000E-01 1.25000000E-01
-3.75000000E-01 -3.75000000E-01 1.25000000E-01
3.75000000E-01 3.75000000E-01 3.75000000E-01
-3.75000000E-01 3.75000000E-01 3.75000000E-01
kptrlatt 4 0 0 0 4 0 0 0 4
kptrlen 2.22880057E+01
lpawu 2 2 1
macro_uj 1
P mkmem 10
natom 4
nband 26
natvshift 5
ndtset 5
ngfft 30 30 30
ngfftdg 40 40 40
nkpt 10
nspden 2
nsppol 2
nsym 12
ntypat 3
occ 1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
0.000000 0.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
1.000000 1.000000 1.000000 1.000000 1.000000 1.000000
0.000000 0.000000
occopt 0
pawecutdg 2.00000000E+01 Hartree
pawujv1 -5.5123983263E-03 Hartree
pawujv2 -3.6749322176E-03 Hartree
pawujv3 -1.8374661088E-03 Hartree
pawujv4 1.8374661088E-03 Hartree
pawujv5 3.6749322176E-03 Hartree
prtden 0
prtebands 0
prteig 0
prtgsr 0
prtwf 0
rprim 5.0000000000E-01 5.0000000000E-01 1.0000000000E+00
1.0000000000E+00 5.0000000000E-01 5.0000000000E-01
5.0000000000E-01 1.0000000000E+00 5.0000000000E-01
shiftk 5.00000000E-01 5.00000000E-01 5.00000000E-01
spgroup 166
spinat 0.0000000000E+00 0.0000000000E+00 3.0000000000E+00
0.0000000000E+00 0.0000000000E+00 -3.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten1 1.4248393785E-01 1.4248393785E-01 1.4248393785E-01
-4.8558537555E-04 -4.8558537555E-04 -4.8558537555E-04
strten2 1.4247967561E-01 1.4247967561E-01 1.4247967561E-01
-4.8629968488E-04 -4.8629968488E-04 -4.8629968489E-04
strten3 1.4247518212E-01 1.4247518212E-01 1.4247518212E-01
-4.8713661212E-04 -4.8713661212E-04 -4.8713661212E-04
strten4 1.4246471396E-01 1.4246471396E-01 1.4246471396E-01
-4.8947873252E-04 -4.8947873252E-04 -4.8947873252E-04
strten5 1.4245835000E-01 1.4245835000E-01 1.4245835000E-01
-4.9122927842E-04 -4.9122927842E-04 -4.9122927842E-04
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
tolvrs 1.00000000E-09
typat 1 2 3 3
usepawu 1
useylm 1
wtk 0.03125 0.09375 0.09375 0.09375 0.09375 0.18750
0.18750 0.09375 0.03125 0.09375
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
4.1699164191E+00 4.1699164191E+00 4.1699164191E+00
2.0849582095E+00 2.0849582095E+00 2.0849582095E+00
6.2548746286E+00 6.2548746286E+00 6.2548746286E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
7.8800000000E+00 7.8800000000E+00 7.8800000000E+00
3.9400000000E+00 3.9400000000E+00 3.9400000000E+00
1.1820000000E+01 1.1820000000E+01 1.1820000000E+01
xred 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 5.0000000000E-01
2.5000000000E-01 2.5000000000E-01 2.5000000000E-01
7.5000000000E-01 7.5000000000E-01 7.5000000000E-01
znucl 28.00000 28.00000 8.00000
================================================================================
- Timing analysis has been suppressed with timopt=0
================================================================================
Suggested references for the acknowledgment of ABINIT usage.
The users of ABINIT have little formal obligations with respect to the ABINIT group
(those specified in the GNU General Public License, http://www.gnu.org/copyleft/gpl.txt).
However, it is common practice in the scientific literature,
to acknowledge the efforts of people that have made the research possible.
In this spirit, please find below suggested citations of work written by ABINIT developers,
corresponding to implementations inside of ABINIT that you have used in the present run.
Note also that it will be of great value to readers of publications presenting these results,
to read papers enabling them to understand the theoretical formalism and details
of the ABINIT implementation.
For information on why they are suggested, see also https://docs.abinit.org/theory/acknowledgments.
-
- [1] Facilities and practices for linear response Hubbard parameters U and J in Abinit.
- L. MacEnulty, M. Giantomassi, B. Amadon, G.-M. Rignanese and D.D. O'Regan Electron. Struct. 6 037003 (2024).
- Comment: to be cited in case the Hubbard U or Hund's J are calculated using the lrUJ utility
- or the renovated functionalities of UJdet, i.e., macro_uj>0.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#MacEnulty2024
-
- [2] Gamma and beta cerium: DFT+U calculations of ground-state parameters.
- B. Amadon, F. Jollet and M. Torrent, Phys. Rev. B 77, 155104 (2008).
- Comment: DFT+U calculations, usepawu/=0. Strong suggestion to cite this paper.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#amadon2008a
-
- [3] Implementation of the Projector Augmented-Wave Method in the ABINIT code.
- M. Torrent, F. Jollet, F. Bottin, G. Zerah, and X. Gonze Comput. Mat. Science 42, 337, (2008).
- Comment: PAW calculations. Strong suggestion to cite this paper.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#torrent2008
-
- [4] Libxc: A library of exchange and correlation functionals for density functional theory.
- M.A.L. Marques, M.J.T. Oliveira, T. Burnus, Computer Physics Communications 183, 2227 (2012).
- Comment: to be cited when LibXC is used (negative value of ixc)
- Strong suggestion to cite this paper.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#marques2012
-
- [5] Abinit 2025: New Capabilities for the Predictive Modeling of Solids and Nanomaterials.
- J. Chem. Phys. 163, 164126 (2025).
- M.J. Verstraete, J. Abreu, G. Allemand, B. Amadon, G. Antonius,
- M. Azizi, L. Baguet, C. Barat, L. Bastogne, R. Bejaud, J.-M. Beuken,
- J. Bieder, A. Blanchet, F. Bottin, J. Bouchet, J. Bouquiaux, E. Bousquet,
- J. Boust, F. Brieuc, V. Brousseau-Couture, N. Brouwer, F. Bruneval,
- A. Castellano, E. Castiel, J.-B. Charraud, J. Clerouin, M. Cote, C. Duval,
- A. Gallo, F. Gendron, G. Geneste, Ph. Ghosez, M. Giantomassi, O. Gingras,
- F. Gomez-Ortiz, X. Gonze, F. Goudreault, A. Gruneis, R. Gupta, B. Guster,
- D.R. Hamann, Xu He, O. Hellman, N. Holzwarth, F. Jollet, P. Kestener,
- I.-M. Lygatsika, O. Nadeau, L. MacEnulty, E. Mara
- Comment: the sixth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for J. Chem. Phys.
- is available at
- https://github.com/abinit/abinit_assets/tree/main/abinit_package/papers/ABINIT25.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#verstraete2025
-
- [6] The Abinit project: Impact, environment and recent developments.
- Computer Phys. Comm. 248, 107042 (2020).
- X.Gonze, B. Amadon, G. Antonius, F.Arnardi, L.Baguet, J.-M.Beuken,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, N.Brouwer, F.Bruneval,
- G.Brunin, T.Cavignac, J.-B. Charraud, Wei Chen, M.Cote, S.Cottenier,
- J.Denier, G.Geneste, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, Xu He, N.Helbig, N.Holzwarth, Y.Jia, F.Jollet,
- W.Lafargue-Dit-Hauret, K.Lejaeghere, M.A.L.Marques, A.Martin, C.Martins,
- H.P.C. Miranda, F.Naccarato, K. Persson, G.Petretto, V.Planes, Y.Pouillon,
- S.Prokhorenko, F.Ricci, G.-M.Rignanese, A.H.Romero, M.M.Schmitt, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, G.Zerah and J.W.Zwanzig
- Comment: the fifth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at
- https://github.com/abinit/abinit_assets/tree/main/abinit_package/papers/ABINIT20.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2020
-
- [7] The Abinit project: Impact, environment and recent developments.
- Computer Phys. Comm. 248, 107042 (2020).
- X.Gonze, B. Amadon, G. Antonius, F.Arnardi, L.Baguet, J.-M.Beuken,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, N.Brouwer, F.Bruneval,
- G.Brunin, T.Cavignac, J.-B. Charraud, Wei Chen, M.Cote, S.Cottenier,
- J.Denier, G.Geneste, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, Xu He, N.Helbig, N.Holzwarth, Y.Jia, F.Jollet,
- W.Lafargue-Dit-Hauret, K.Lejaeghere, M.A.L.Marques, A.Martin, C.Martins,
- H.P.C. Miranda, F.Naccarato, K. Persson, G.Petretto, V.Planes, Y.Pouillon,
- S.Prokhorenko, F.Ricci, G.-M.Rignanese, A.H.Romero, M.M.Schmitt, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, G.Zerah and J.W.Zwanzig
- Comment: a global overview of ABINIT, with focus on selected capabilities .
- Note that a version of this paper, that is not formatted for J. Chem. Phys.
- is available at
- https://github.com/abinit/abinit_assets/tree/main/abinit_package/papers/ABINIT20_JPC.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#romero2020
-
- Proc. 0 individual time (sec): cpu= 106.1 wall= 106.2
================================================================================
Calculation completed.
.Delivered 20 WARNINGs and 6 COMMENTs to log file.
+Overall time at end (sec) : cpu= 106.1 wall= 106.2
You will find all information related to the calculation of the U parameter between the
calculate U, (J) flags. The first information printed out is as follows:
*********************************************************************
************************ Linear Response U ************************
Info printed for perturbed atom: 1
Perturbations Occupations
--------------- -----------------------------
alpha [eV] Unscreened Screened
--------------- -----------------------------
0.0000000000 8.6380190174 8.6380190174
-0.1500000000 8.6964896369 8.6520721437
Scalar response functions:
Chi0 [eV^-1]: -0.86623
Chi [eV^-1]: -0.09369
The scalar U from the two-point regression scheme is 9.51936 eV.
*********************************************************************
*********************************************************************
Here, ujdet lists out the perturbation strengths and their corresponding unscreened and
screened occupations. (When calculating the Hund’s J, Occupations will be replaced by
Magnetizations, and alpha will be replaced by beta.) From
here, the scalar response functions \(\chi\) and \(\chi_0\) are printed out, followed by the scalar
definition of U in units of eV. By scalar, we mean that the response functions are treated as
scalars, and thus the U printed here is informed only by the occupational responses on the
perturbed atom.
The lines starting with URES, by contrast, report the U as a function of its inverted (and charge bath augmented) response matrices:
URES ii nat r_max U(J)[eV] U_ASA[eV] U_inf[eV]
URES 1 1 0.00000 2.37984 1.91514 1.85257
URES 2 8 11.14400 8.34413 6.71480 6.49545
URES 3 27 12.45940 9.16724 7.37718 7.13619
URES 4 64 22.28800 9.37065 7.54088 7.29454
URES 5 125 24.28780 9.44321 7.59926 7.35102
URES 6 216 33.43200 9.47529 7.62508 7.37599
These values of U are computed using the extrapolation procedure proposed in [Cococcioni2005]. In this work, it is shown that using a two atom supercell for the DFT calculation and an extrapolation procedure can yield an estimation of the value of U. More precise values can be and often are obtained by running linear response DFT calculations on larger and larger supercells. Doing so have the added benefit of isolating the perturbed subspace from its periodic images.
The column nat indicates how many atoms were involved in the extrapolated supercell,
and r_max indicates the maximum distance of the perturbed atom from its periodic
images. The column U(J) reports in eV the calculated values of the Hubbard parameters that should
then be applied via the Hubbard functionals. U_ASA and U_inf are estimates
of U for more extended projectors; they are related to the charge population analysis conducted
under the PAW method and controlled by variable dmatpuopt. An numerical analysis of the effect of
dmatpuopt choice is conducted in Ref. [MacEnulty2023].
This is the information printed out for one perturbation of strength pawujv; five further
perturbations are conducted, for each of which the same information is printed in the output file.
You should have five *_LRUJ.nc files in your directory, which we will use as input for
the lruj post-processing utility to determine a better estimate of the U parameter.
4.3. Execution of the lruj post-processinng utility¶
Once the _LRUJ.nc files are printed, execute the lruj utility (found in the same
directory as the abinit executable) with the following command.
lruj *_LRUJ.nc > tlruj_lruj.out
It should take less than a second to run. If the lruj utility runs successfully, the resulting
output file, tlruj_lruj.out, should look like this:
.Version 10.6.4.5 of LRUJ, released Dec 2025.
.(MPI version, prepared for a x86_64_linux_gnu13.2 computer)
.Copyright (C) 1998-2026 ABINIT group .
LRUJ comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Wed 4 Mar 2026.
- ( at 12h43 )
Number of perturbations detected: 5
Including unperturbed state, we have 6 data points.
Standard determination of the Hubbard U parameter.
Maximum degree of polynomials analyzed: 3
NOTE: Unlike the ujdet utility, lruj treats the
response functions as scalars, not matrices!
See lruj tutorial for more information.
*************************************************************************************************
************************************** Linear Response U **************************************
Total number of atoms: 4
Index of perturbed atom: 1
Value of macro_uj: 1
Value of dmatpuopt: 3
Mixing constant factored out of Chi0: 0.450
Percentage of AE orbital within the PAW sphere of perturbed subspace: 90.46723%
Perturbations Occupations
--------------- -----------------------------
alpha [eV] Unscreened Screened
--------------- -----------------------------
-0.1500000000 8.6964981863 8.6520721987
-0.1000000000 8.6747201546 8.6474287205
-0.0500000000 8.6560314451 8.6427490198
0.0000000000 8.6380182458 8.6380182458
0.0500000000 8.6192514500 8.6332174908
0.1000000000 8.5980252049 8.6283148758
RMS Errors
---------------------------------------
Regression Chi0 [eV^-1] Chi [eV^-1] U [eV] | Chi0 [eV^-1] Chi [eV^-1] U [eV]
--------------------------------------------------------|---------------------------------------
Linear: -0.8594085 -0.0949435 9.3689919 | 0.0023925 0.0000878 0.0102652
Quadratic: -0.8574668 -0.0955792 9.2963067 | 0.0023777 0.0000129 0.0035293
Cubic: -0.8007861 -0.0952726 9.2474189 | 0.0001546 0.0000015 0.0002899
*************************************************************************************************
*************************************************************************************************
Linear Response UJ (LRUJ) program complete. Live long and prosper. ~LMac
--- !LRUJ_Abipy_Plots
natom: 4
ndata: 6
pawujat: 1
macro_uj: 1
diem_token: diemix
diem: 4.50000000E-01
chi0_coefficients_degree1: [ 8.63742244E+00, -3.86733840E-01, ]
chi_coefficients_degree1: [ 8.63792651E+00, -9.49434730E-02, ]
chi0_coefficients_degree2: [ 8.63730593E+00, -3.85860059E-01, 1.74756283E-02, ]
chi_coefficients_degree2: [ 8.63801126E+00, -9.55791520E-02, -1.27135811E-02, ]
chi0_coefficients_degree3: [ 8.63801773E+00, -3.60353751E-01, -1.60475359E-01, -2.37267982E+00, ]
chi_coefficients_degree3: [ 8.63801982E+00, -9.52726489E-02, -1.48519747E-02, -2.85119145E-02, ]
...
This specific calculation looks at the Hubbard U parameter (macro_uj 1) using results from five (5) perturbations, the strengths of which are listed in the first table alongside the corresponding subspace occupations, both unscreened (for \(\chi_0\), harvested during the first SCF cycle, immediately after the perturbation is applied but before the Hamiltonian is updated) and screened (for \(\chi\), harvested from the SCF converged subspaces occupancies).
Perturbations Occupations
--------------- -----------------------------
alpha [eV] Unscreened Screened
--------------- -----------------------------
-0.1500000676 8.6964981921 8.6520721998
-0.1000000451 8.6747201574 8.6474287212
-0.0500000225 8.6560314462 8.6427490201
0.0000000000 8.6380182458 8.6380182458
0.0500000225 8.6192514478 8.6332174978
0.1000000451 8.5980252003 8.6283148749
The last table gives the values for \(\chi_0\), \(\chi\), the Hubbard U, and their RMS errors in units of eV, for all polynomial regressions up to degree 3 (cubic).
RMS Errors
---------------------------------------
Regression Chi0 [eV^-1] Chi [eV^-1] U [eV] | Chi0 [eV^-1] Chi [eV^-1] U [eV]
--------------------------------------------------------|---------------------------------------
Linear: -0.8594082 -0.0949434 9.3689968 | 0.0023925 0.0000878 0.0029335
Quadratic: -0.8574665 -0.0955791 9.2963113 | 0.0023777 0.0000129 0.0027762
Cubic: -0.8007858 -0.0952726 9.2474280 | 0.0001546 0.0000015 0.0001937
One has the option of calculating higher order polynomials, up to degree \(n–2\) for n points. This
is done by appending the degree option –d followed by an integer to the command line.
For example, for polynomial regressions up to degree 4 of the above calculation with 6 data points,
try bashing
lruj *_LRUJ.nc --d 4 > tlruj_lruj_d4.out
to get the following output:
RMS Errors
---------------------------------------
Regression Chi0 [eV^-1] Chi [eV^-1] U [eV] | Chi0 [eV^-1] Chi [eV^-1] U [eV]
--------------------------------------------------------|---------------------------------------
Linear: -0.8594082 -0.0949434 9.3689968 | 0.0023925 0.0000878 0.0029335
Quadratic: -0.8574665 -0.0955791 9.2963113 | 0.0023777 0.0000129 0.0027762
Cubic: -0.8007858 -0.0952726 9.2474280 | 0.0001546 0.0000015 0.0001937
Degree 4 : -0.8049062 -0.0952279 9.2587427 | 0.0000790 0.0000003 0.0000981
Note
The lruj program will only calculate polynomials up to degree \(n–2\) for n data points; so, by
default, if one reads in only two _LRUJ.nc files, the maximum polynomial regression
conducted will be linear; three _LRUJ.nc files means maximum degree is quadratic,
and so on. Other command line options for the lruj utility include –version and
–help.
The values in eV of the Hubbard U parameter according to each regression are found in column four. To assess which one is best, you’ll want to use the RMS errors in column seven. Furthermore, you can import the perturbation/occupation table into Excel (or any other choice of graphical utility) to visualize the fits. The following graph visualizes our data in Mathematica.
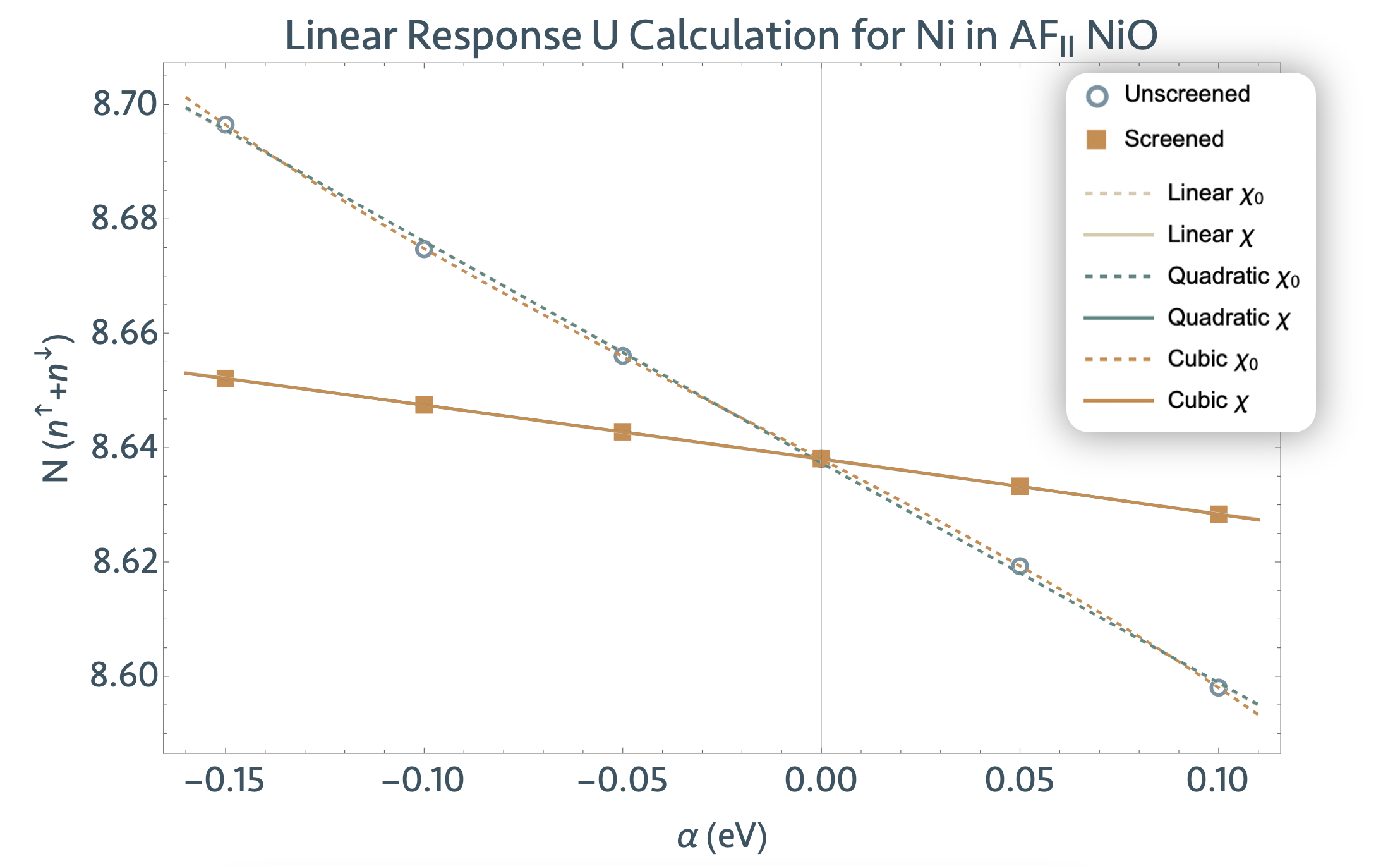
Based on this information, one could argue the quadratic fit is sufficient. Thus, we get a first-principles Hubbard U value of 9.296 ± 0.003 eV for the Ni 3d subspace in a four-atom cell of AF2 NiO. This is much larger than values for NiO reported in the literature values, which can be attributed to the poor ecut and ngkpt sampling needed to speed up this tutorial. Furthermore, this indicates that these parameters must be converged with respect to supercell size in order to isolate the perturbed subspace from its periodic images.
Repeating Steps 1-3 with macro_uj set to 4 will give us the Hund’s J parameter for the same Ni 3d subspaces. We do this now but more precisely, doubling the value of ecut and lowering the tolerance, etc. When we plot the linear response data, it becomes more obvious why more scruitnous run parameters are necessary and why polynomials of higher order are needed to perform an accurate regression:
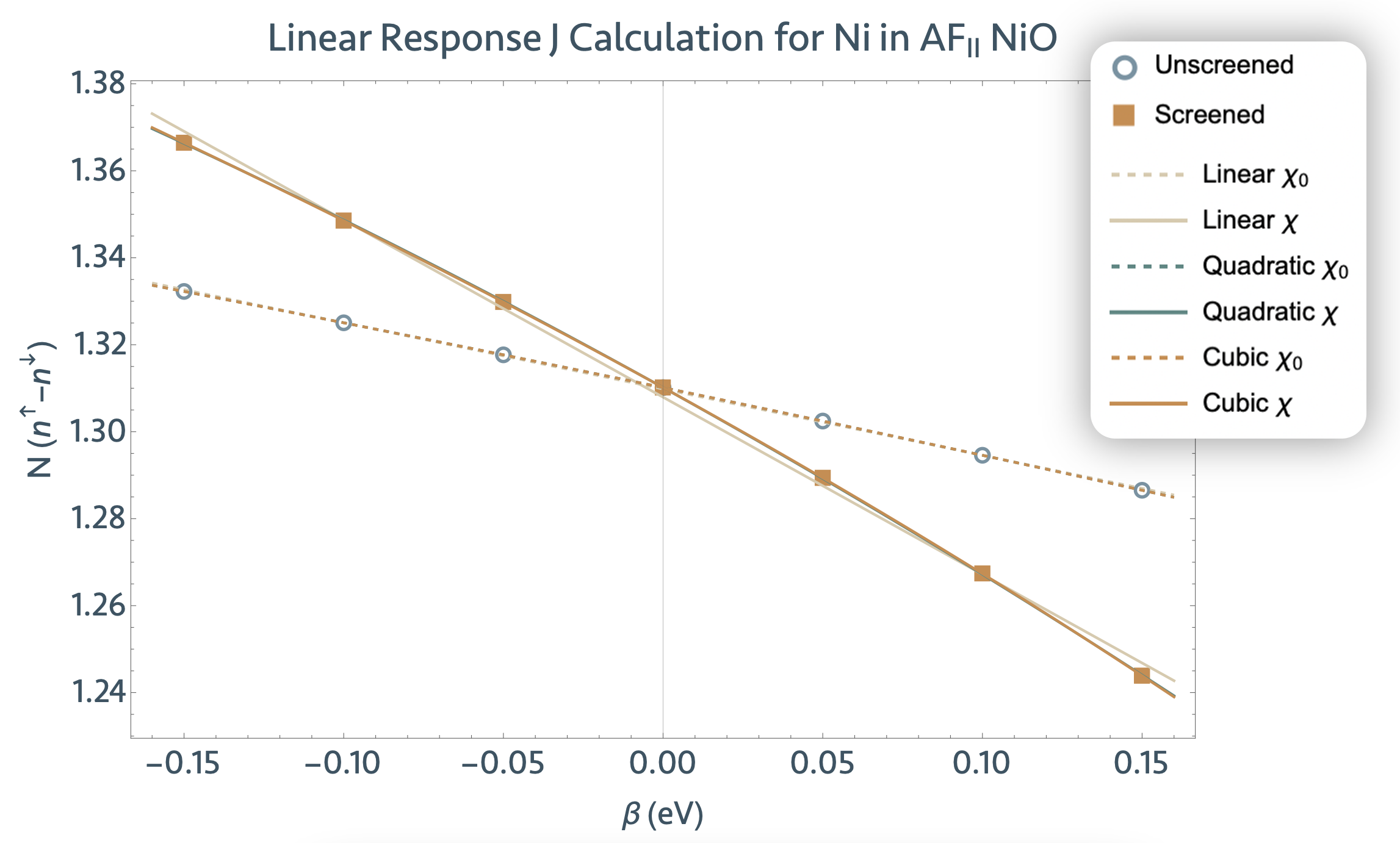
Here, we see that the quadratic fit, at minimum, sufficiently fits the data, yielding a Hund’s J value of 0.4994 ± 0.0004 eV for the Ni 3d subspace in a four-atom cell NiO. Keep in mind, however, that these parameters MUST be converged with respect to supercell size in order to isolate the perturbed subspace from its periodic images.